







 本文详细介绍了ChIP-seq数据分析中的Peak文件格式,包括NarrowPeaks, BroadPeaks和GappedPeaks,并解析了MACS2软件的peakcalling过程和输出文件。此外,还概述了常见的测序文件格式如BAM, SAM, CRAM以及fastq和fasta,以及如何将GTF文件转换为BED格式。
本文详细介绍了ChIP-seq数据分析中的Peak文件格式,包括NarrowPeaks, BroadPeaks和GappedPeaks,并解析了MACS2软件的peakcalling过程和输出文件。此外,还概述了常见的测序文件格式如BAM, SAM, CRAM以及fastq和fasta,以及如何将GTF文件转换为BED格式。
生信搬运工-03
文章目录
一、Peak文件介绍
在进行peak calling分析时,经常会接触到以下3种peak格式
narrow peaks format
broad peaks fotmat
gapped peaks format
peak被定义为基因组上一段reads富集的区域,核心信息是在染色体上的起始和终止位置,除此之外,还有软件对于该peak区域的打分,比如常见的pvalue, qvalue, fold_enrichment等值。
和基因组比对信息用BAM格式来存储类似,为了标准化不同peak calling软件的输出,特意制定了以上3种数据格式。这三种格式本质上都是bed文件,只不过列数不太类似。
1. Narrow Peaks Format
该格式又称之为point-source peaks format, macs2默认输出就是这种格式,是一种BED6+4的格式,列数为10列,可以上传到UCSC浏览。该文件可以直接加载到UCSC基因组浏览器。示意如下
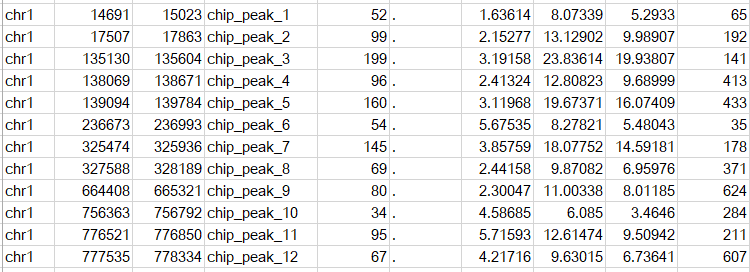
前四列分别代表chrom, chromStart, chromEnd, name, 用于描述peak区间和名称,注意bed格式中起始位置从0开始计数。
第五列score,在macs2的输出结果中为int(-10*log10qvalue),
第六列strand,,在macs2的输出结果中为 . ,
第七列signalvalue,通常使用fold_enrichment的值,
第八列pvalue,在macs2的输出结果中为-log10(pvalue),
第九列qvalue,在macs2的输出结果中为-log10(qvalue),
第十列peak,在macs2的输出结果中为peak的中心,即summit距离peak起始位置的距离。
2. Broad Peaks Format
这种格式就是在narrow peaks format的基础上丢掉了最后一列的信息,为BED6+3的格式, 列数为9列。
3. Gapped Peaks Format
前两种格式都是由于描述连续的peak区间,适用于DNA水平上的富集区域信息的存储,比如chip_seq, ATAC_seq鉴定到的peak区间,而gapped peaks format用于描述非连续的peak区间,这里的非连续通常指的是在peak的区间内会包含多个exon区域,适用于RNA水平上的富集区域信息的存储,比如m6A_seq鉴定到的peak区间。
该格式在BED12的基础上进行延伸,演变为BED12+3的格式,列数为15列,每列的含义示意如下

前6列的含义和上述两种peak格式完全相同,后3列的含义和broad peak完全相同,为了专区表示peak区间内包含的exon信息,借鉴转录本的BED12格式,引入了以下6列
thickStart
thickEnd
itemRgb
blockCount
blockSizes
blockStarts
thickStart和thickEnd有点类似转录本中CDS的起始和终止位置,在存储peak信息时,通常的做法是将这两列的值和chromStart和chromEnd的值设置成相同的,itemRgb是一个RGB颜色值,比如255,0,0, 如果没有对应的颜色信息,则用0来表示。
blockCount代表该peak区间包含的exon的个数,blockSizes代表每个exon区间的长度,多个exon用逗号连接,blockStarts代表每个exon区间在基因组上的起始位置,多个exon用逗号连接。
二、Macs2 peak calling实战
MACS是一款最为流行的peak calling软件,最初是针对转录因子的chip数据来设计的,在最新版本中,也添加了对组蛋白修饰的适配。目前最新版本为v2.0,官网如下
https://github.com/taoliu/MACS
在2.0版本中提供了以下多个子命令,每个子命令和对应的功能描述如下
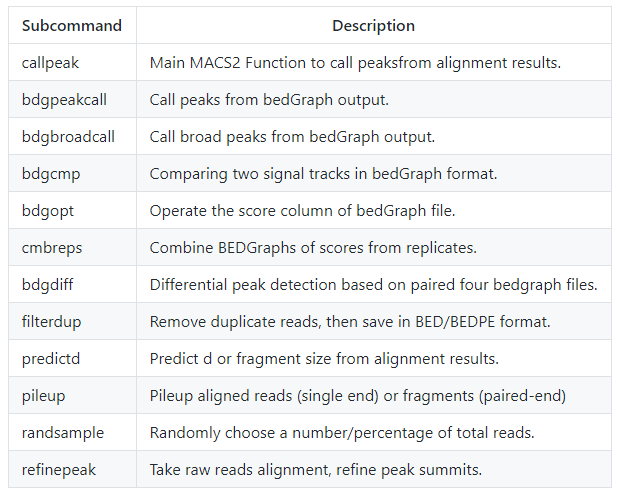
本文主要介绍macs2最经典的使用场景peak calling, 基本用法如下
macs2 callpeak \
-t ip.bam \
-c input.bam \
--outdir out_dir \
-n chip \
-g hs
-t参数指定抗体处理的样本,-c指定input样本,值得一提的是,macs支持多种格式的输入文件,除了上述代码中使用的bam格式外,还支持SAM/BED格式。
–outdir指定输出结果的目录,-n参数指定输出文件名的前缀,-g参数指定基因组的有效大小,在NGS数据中,测序reads在基因组上的覆盖度并不是100%, 而且有些重复区域的比对信息是不可信的,剩下的能够利用的区域通常只占整个基因组区域的70%到90%,这个区域的大小就是有效大小,对于常见的物种,程序内置了有效大小,我们只需要指定物种的缩写即可,如
- hs:2.7e9
- mm: 1.87e9
- ce: 9e7
- dm: 1.2e8
chip_model.r
chip_peaks.narrowPeak
chip_peaks.xls
chip_summits.bed
model.r是一个可执行的R脚本,通过以下代码可以产生一个PDF的输出文件
Rscript chip_model.r
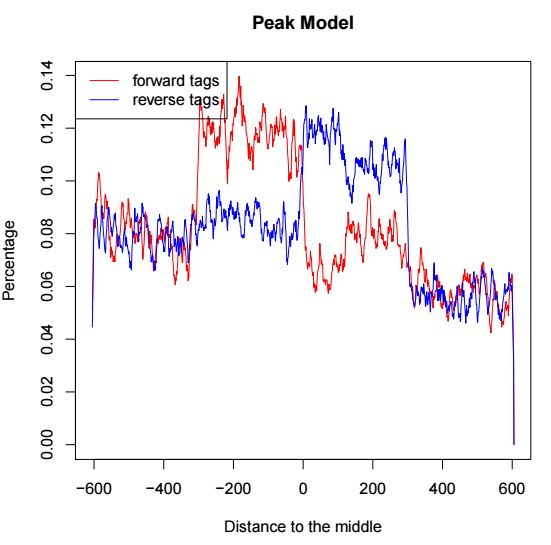
第一页表示peak邻近区间正负链测序分布,用于评估d这个参数值,示意如下
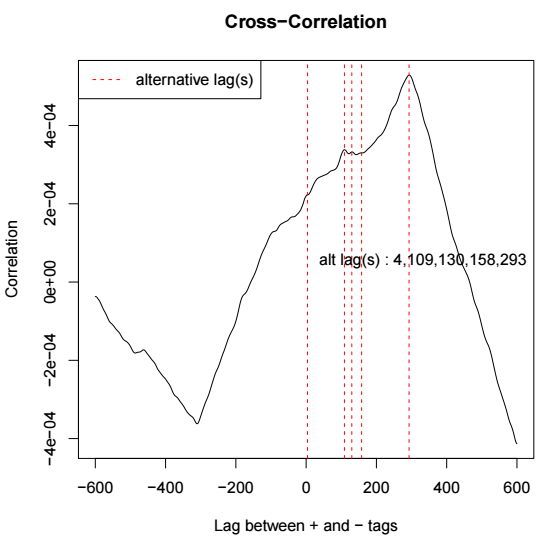
第二页是cross-correlation分析的结果,示意如下
后缀为xls的文件是peak的输出结果,开头的是注释信息,显示了软件调用的具体命令和参数设置,便于核查;其他的行记录了peak的区间信息,这里的起始位置采用的是从1开始计数的方式。
后缀为narrowpeak的文件是一个BED格式的文件,bed格式中起始位置从0开始计数,xls文件的起始位置-1才和bed文件一致
后缀为bed的文件为peak中心,即summit对应的bed文件,
以上就是macs2 peak calling的基本用法,更多详细的参数和用法请参考官方文档。
三、测序常见文件格式
bam/sam/cram
sam: SAM是一种序列比对格式标准, 由sanger制定,是以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结
bam: sam文件的二进制压缩文件,可使用samtools工具查看。由于其运行速度快,所以常常使用bam而不是sam。(B取自binary)
当测序得到的fastq文件map到基因组之后,我们通常会得到一个sam或者bam为扩展名的文件。SAM的全称是sequence alignment/map format。
CRAM: fastq/fasta
fastq: 一种存储了生物序列(通常是核酸序列)以及相应的质量评价的文本格式。
fasta: 是一种基于文本用于表示核酸序列或多肽序列的格式。其中核酸或氨基酸均以单个字母来表示,且允许在序列前添加序列名及注释。
##fastq to fasta
zcat input.fastq.gz | awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' > output.fa
less -S SRR5586990.fastq| paste - - - -|cut -f 1-2|tr '\t' '\n'|tr '@' '>' |less -S > SRR5586990.fasta
##统计序列条数(reads)
zcat *R1.fq.gz |grep '@'| wc –l或者zcat *.fastq.gz | awk 'NR%4==2{c++} END{print c}'
pigz -dc input.fastq.gz | awk 'NR%4==2{c++} END{print c}' #速度快10倍
##GC含量及碱基数目
perl -ne 'if($.%4){chomp;$count_G=$count_G+($_=~tr/G//);$count_C=$count_C+($_=~tr/C//);$cur_length=length($_);$total_length+=$cur_length;}END{print qq{total count is $total_length bp\nGC%:},($count_G+$count_C)/$total_length,qq{\n} }' input.fq
gtf/gff3/GenePred
以gencode下人类的gtf文件为例,手册页https://www.gencodegenes.org/pages/data_format.html有详细的格式说明。
GENCODE和Ensembl数据库均提供具有基因组坐标的GTF / GFF3文件,但不提供BED文件。
gtf to bed
grep -P "\tgene\t" gencode.xxxxxx.gtf | \ #第三列筛选gene,(或者transcripts之类的)
cut -f1,4,5,7,9 | \ #提取所需列
sed 's/[[:space:]]/\t/g' | \ #空格替换制表符
sed 's/[;|"]//g' | \ #去掉;”
awk -F $'\t' 'BEGIN { OFS=FS } { print $1,$2-1,$3,$6,".",$4,$10,$12,$14 }' | \ #bed文件起始位置从0开始,需要减一,这里你可以选择你需要的列,一般前三列加个gene symbol那一列就够了。
>genecode_by_gene.txt
在R中操作
rtracklayer包(R的基因组浏览器接口)
将gtf在R中转为数据框进行筛选。[或者直接对GenomicRanges对象进行操作]
gtf <- rtracklayer::import("xxxxxxxx.gtf")
library(tidyverse)
gtf %>% as_tibble() %>%
filter(type == "gene") %>%
mutate(start = start -1) %>%
select(1:3,gene_id,gene_name) %>%
write_tsv("xxx.bed")
gtf to gff3,GenePred
总结
原文链接:https://blog.csdn.net/weixin_43569478/article/details/108079436
原文链接:https://blog.csdn.net/weixin_43569478/article/details/108079439
原文链接:https://blog.csdn.net/weixin_45433910/article/details/109149877

















 738
738

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









