







这篇文章分为三个部分,第一部分是基本概念介绍
第二部分是官方示例代码的重现
第三部分是用自己的数据重现代码
基本概念介绍
Scanpy 和 Seurat 基本上完全一样,Scanpy 构建的对象叫做 AnnData 对象,他的数据存储是以4 个模块存储(如下图)
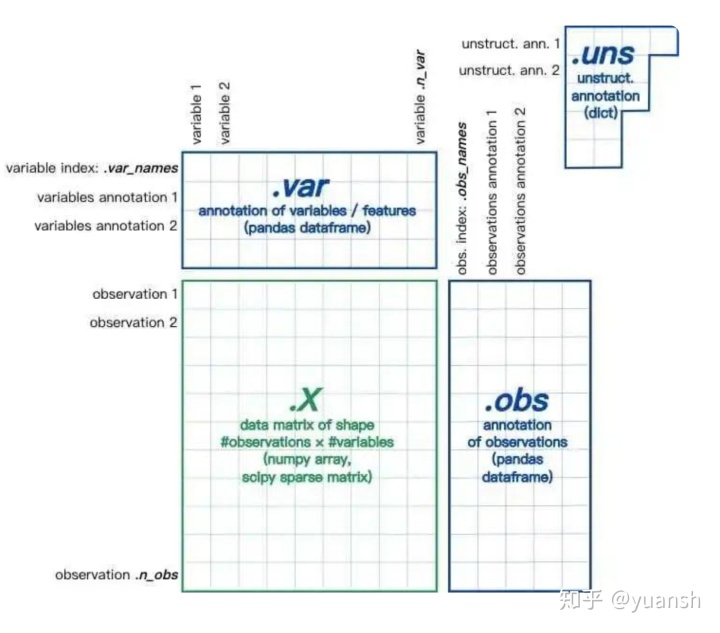
其中X对象为count 矩阵。这里要注意一下,它和 R 语言的不同,Scanpy 中的行为样本,列为基因。这也和 python 的使用习惯相关
- X 对象为count 矩阵,与 seurat 对象是转置关系
- obs 存储的是 seurat 对象中的 meta.data 矩阵
- var 存储的是基因(特征)的信息
- uns 存储的是后续添加的非结构信息
官方示例代码
import scanpy as sc
import os
import math
import itertools
import warnings
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
%matplotlib inline
%config InlineBackend.figure_format = 'svg'
warnings.filterwarnings("ignore")
plt.rc('font',family='Times New Roman')
my_colors = ["#1EB2A6","#ffc4a3","#e2979c","#F67575"]
sc.settings.verbosity = 3 # 输出提示信息
# ?sc.settings.verbosity
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')# 设置输出图像格式
results_file = 'write/pbmc3k.h5ad' # 存储分析结果
scanpy==1.6.0 anndata==0.7.5 umap==0.4.6 numpy==1.19.2 scipy==1.4.1 pandas==1.1.3 scikit-learn==0.23.2 statsmodels==0.12.0这里的读取文件的方式和R语言构造seurat对象基本一样 (按照官网分类有12中读取方式)
下面主要介绍两种方法
第一种方法是,文件下面要有3个初始文件包括:
- barcord
- genes
- matrix
然后使用输sc.read_10_mtx读取
第二种方法是直接构建AnnData对象
然后分别的将表达矩阵,细胞信息,基因信息读取,代码如下
# 这个是第二种方法
# creat scanpy object
#df = pd.read_csv('processfile/count.csv', index_col=0)
#meta = pd.read_csv('processfile/metadata.csv', index_col=0)
#cellinfo = pd.DataFrame(df.index,index=df.index,columns=['sample_index'])
#geneinfo = pd.DataFrame(df.columns,index=df.columns,columns=['genes_index'])
#sce = sc.AnnData(df, obs=cellinfo, var = geneinfo)
# 这个是第一种读取方法
adata = sc.read_10x_mtx(
'./filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
cache=True)
adata.var_names_make_unique()
adatatips: pytho和R语言有点不同,通常情况下,行为样本, 列为特征
adata.obs.shape # 2700个细胞
adata.var.shape # 32738个基因
adata.to_df().shape # 2700*32738
adata.obs.head()
adata.var.head()
adata.to_df().iloc[0:5,0:5]数据预处理
这里介绍一下scanpy中常用的组件
- pp: 数据预处理
- tl: 添加额外信息
- pl:可视化
统计基因在细胞中的占比并可视化
sc.pl.highest_expr_genes(adata, n_top=20) # 每一个基因在所有细胞中的平均表达量(这里计算了百分比含量)
sc.pp.filter_cells(adata, min_genes=200) # 每一个细胞至少表达200个基因
sc.pp.filter_genes(adata, min_cells=3) # 每一个基因至少在3个细胞中表达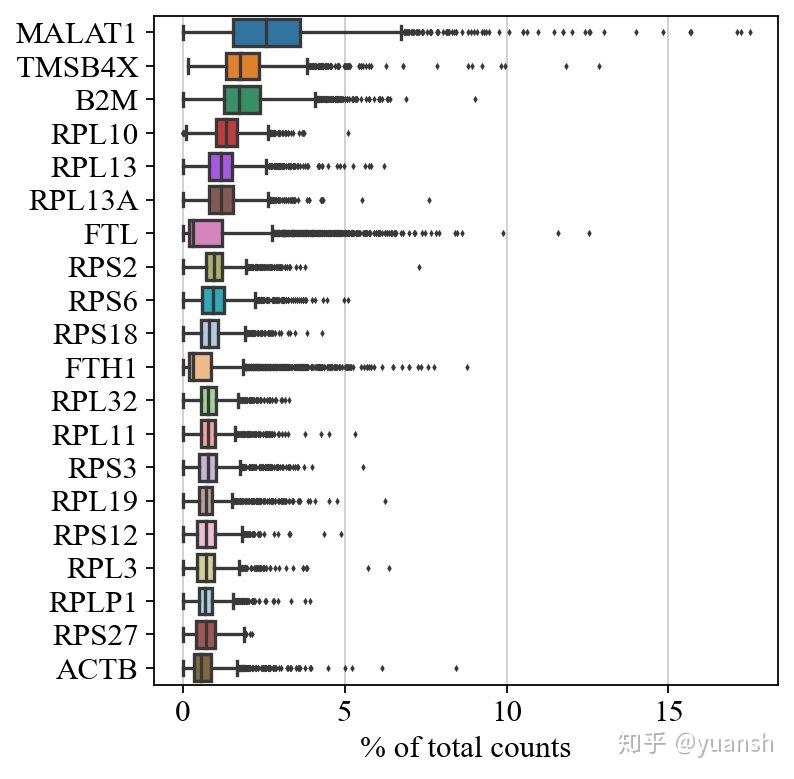
过滤线粒体DNA
str.startswith 不支持正则,如果要使用正则则使用.str.match
sce.var_names[sce.var_names.str.match(r'^MT-')]
sce.var_names[sce.var_names.str.match(r'^RP[SL0-9]')]
sce.var_names[sce.var_names.str.match(r'^ERCC-')]
# 抽取带有MT的字符串
adata.var['mt'] = adata.var_names.str.startswith('MT-')
# 数据过滤
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
# 过滤后可视化(官方文档真的骚到我头皮发麻)
sc.pl.violin(adata, ['n_genes_by_counts'],jitter=0.4)
sc.pl.violin(adata, ['total_counts'],jitter=0.4)
sc.pl.violin(adata, ['pct_counts_mt'],jitter=0.4) 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章















 682
682











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









