







 本文详细介绍了如何使用AutoDockVina进行分子对接,包括从SMILES转换pdb文件,pdb到pdbqt的转换,分子加氢,设置对接盒子,保存配置,以及批量对接操作。此外,还提到了PymOL在绘图和处理分子结构中的作用。
本文详细介绍了如何使用AutoDockVina进行分子对接,包括从SMILES转换pdb文件,pdb到pdbqt的转换,分子加氢,设置对接盒子,保存配置,以及批量对接操作。此外,还提到了PymOL在绘图和处理分子结构中的作用。
完整的代码直接看:D:\Pycharm_workspace\gypsum_dl-1.2.0,这里面问我已经写好了很多功能,包括:
- smiles2pdb:(smiles2pdb.py)
- pdb2pdbqt:(docking/pdb2pdbqt.py)
- 批量docking亲和力计算:(docking/docking_affinity_score.py)
最近看到一个新的对接程序:GitHub - codezhouj/Molecular_Docking
一、得到蛋白质的pdbqt文件:
AutoDock Vina 安装教程:1安装_哔哩哔哩_bilibili
下载地址:https://vina.scripps.edu/wp-content/uploads/sites/55/2020/12/autodock_vina_1_1_2_win32.msi
可视化工具下载:MGLTools:Downloads – mgltools
注意:安装过程中要注意按照视频中的教程将vina.exe复制到这里,否则后面会出错:

附:用PYMOL绘制图片教程:13用PYMOL绘制图片_哔哩哔哩_bilibili
1、去除蛋白质中的小分子
最后用于分子对接的受体的pdbqt文件是要去除小分子的,可以使用pymol去除小分子,从而得到没有小分子的配体文件
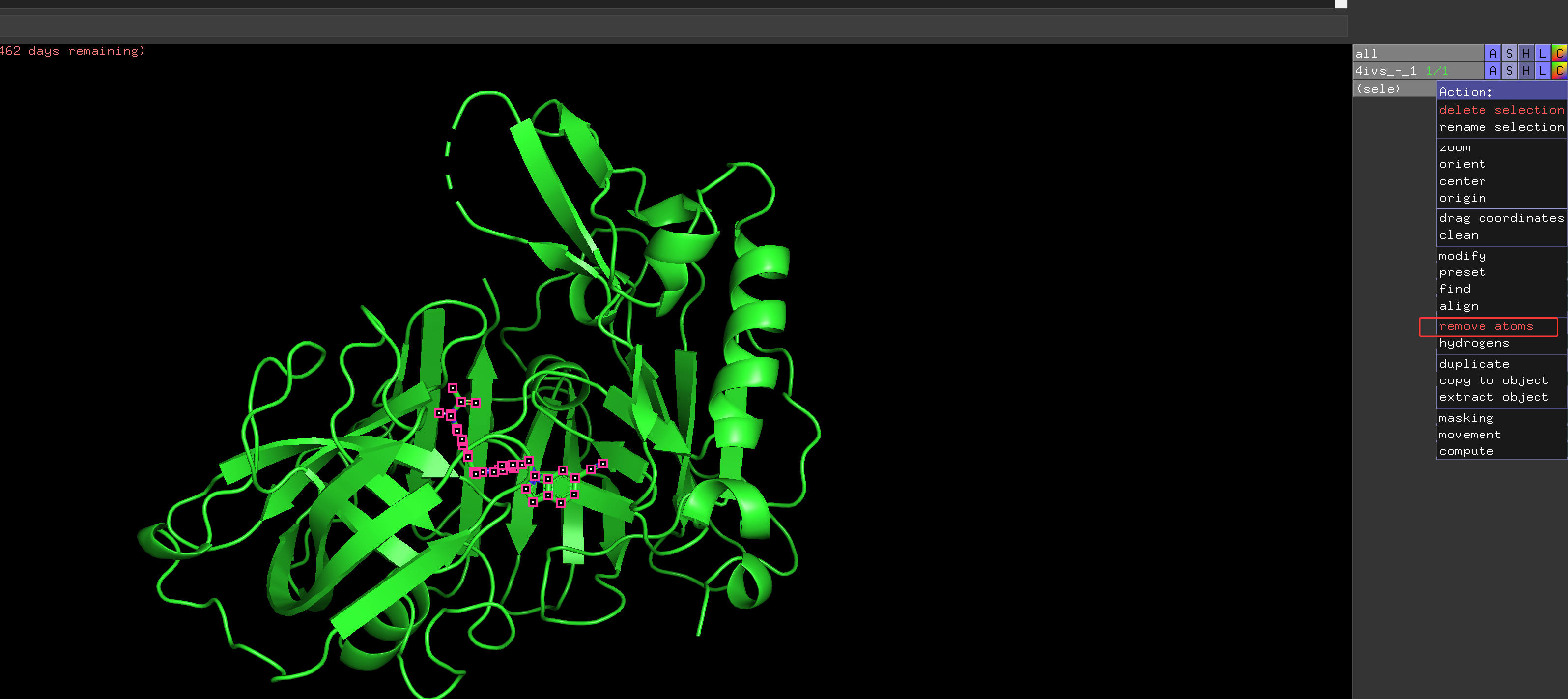
用pymol删除以后然后保存为pdb

一、导入蛋白质pdb文件(去水、加氢)
注意:
最后用于分子对接的受体的pdbqt文件是要去除小分子的,可以使用pymol去除小分子,从而得到没有小分子的配体文件

发现有很多小红点(水分子)【也有可能没有】

1、去掉水分子

2、加氢

然后点击“ok”就可以了

3、将该pdb选为受体:


4、 点击确定保存pdbqt文件

保存后出现该受体的pdbqt文件:

就变成这样了

5、然后将该受体删除

二、小分子操作(读取-->加氢-->导出为pdbqt-->删除)
0、怎么得到小分子?
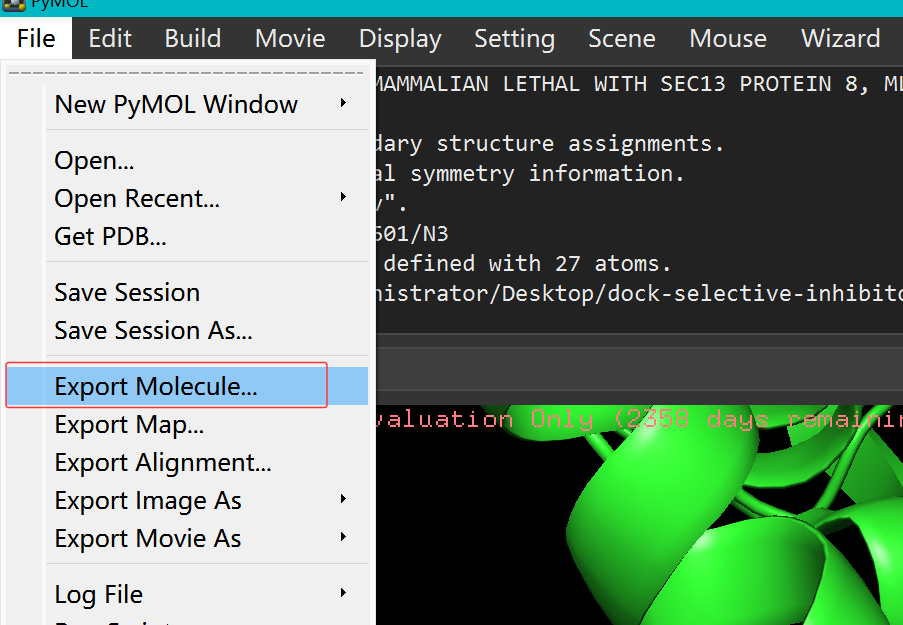
注意:这里的小分子建议直接用pymol打开的受体pdb文件,然后选中小分子,复制到新的pdb文件(因为方便后面盒子的设置)

点击小分子

file --> export molrcule-->新的小分子pdb文件(不要导出成其他格式的了)
1、 打开小分子,然后加氢和上面对配体的加氢操作一样
继续回到autodock:

加氢
2、然后将这个小分子选为配体

点击“YES”
3、选择扭转中心

4、设置扭转键

5、导出为pdbqt文件

6、删除小分子

三、设置盒子,然后存储盒子的信息
1、open前面加氢去水的pdbqt受体文件

2、打开小分子

此时你可以使用不同的表示,从而更清楚的看配体和受体,

这里可以看到,小分子正好在受体的口袋里,这里因为上面我们选择小分子的时候,小分子是直接从受体中选出的

3、 设置盒子

最简单的操作方式(这样盒子正好在小分子的中心):

将盒子罩住蛋白质的活性位点,不需要管是不是罩住了整个蛋白质(这里是个关键,如果你知道这个蛋白质的活性位点、否则你需要用盒子全部罩住这个蛋白质,但是全部罩住可能会导致预测的不准确):

5、去除小分子
如果前面你使用了pymol将小分子和蛋白质分离开了,刚开始打开的也是不带有小分子的蛋白质,那么可以不用执行这一步

6、选中黄色部分的小分子,然后按住鼠标右键将小分子拖出来

7、将选项勾回去,使小分子留在外面

4、保存文件

5、导出config.TXT文件
注意:每次找一个新的受体的盒子的位置的时候需要重新打开一次软件,否则会导致信息(config)无法正确更新

直接就导出到:E:\AutodockVina了
四、单次docking操作
1)打开蛋白质pdbqt文件

2)打开小分子

3) 设置config文件

选择你设置好的config文件

launch就可以了,在命令行中就出现结果了
注意:
这里你导出的config.txt文件可能和你当时盒子设置的参数不一样(例如盒子的大小),这里你需要打开config文件,然后与你的设置比较,然后换成你在“Autodock toolS”中设置的参数才可以
五、批量:
最后将受体的这一行删除就行,因为我们要做的是批量docking

此时我的config.txt是:
receptor = 5ywy.pdbqt
center_x = -44.249
center_y = -42.537
center_z = -1.145
size_x = 15.0
size_y = 15.0
size_z = 15.0
exhaustiveness = 16
num_modes = 9
seed = 2023二、得到smiles(配体)对应的pdbqt + sdf文件(批量)
完整的代码直接看:D:\Pycharm_workspace\gypsum_dl-1.2.0,这里面问我已经写好了很多功能,包括:
smiles2pdb:(smiles2pdb.py)
cd gypsum_dl-1.2.0/
python smiles2pdb_sdf.py --source ligand_smi/image2smiles_validity-Adenosine_A2a_receptor-sample-1k.smi \
--output_folder docking_data/ligand/Adenosine_A2a_receptor/pdb_sdf \
--max_variants_per_compound 1 --add_pdb_output --separate_output_files
三、进行docking
方法一:使用CPU对接( gypsum_dl-1.2.0)
1)pdb2pdbqt:(docking/pdb2pdbqt.py)
python docking/pdb2pdbqt.py --pdb_file docking_data/EP4_ligand/pdb --pdbqt_file docking_data/EP4_ligand/pdbqt
2)使用Uni-Dock进行受体与配体的对接(批量)
批量对接程序在本地:
D:\Pycharm_workspace\gypsum_dl-1.2.0\docking\docking_affinity_score.py
python docking/docking_affinity_score.py --receptor_file docking_data/receptor/3eml/3eml.pdbqt \
--ligand_dir docking_data/ligand/Adenosine_A2a_receptor/pdbqt --config_file docking_data/receptor/3eml/config.txt \
--out_dir docking_data/docking_results
方法二:使用GPU对接( autodock)
https://mapengsen.blog.csdn.net/article/details/129682793
Autodock 分子对接 [2] - 用Autodock Vina / QuickVina2 / QuickVina-W做对接_哔哩哔哩_bilibili



















 934
934

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











