







 本文提出了一种基于深度学习的CPI预测模型PSC-CPI,它通过多尺度序列-结构对比,捕捉蛋白质序列和结构的依赖关系。该模型解决了模态缺失和域转移问题,并通过长度可变的蛋白质增强,增强了模型的泛化能力。模型在没有观察到的化合物或蛋白质情况下仍表现良好。
本文提出了一种基于深度学习的CPI预测模型PSC-CPI,它通过多尺度序列-结构对比,捕捉蛋白质序列和结构的依赖关系。该模型解决了模态缺失和域转移问题,并通过长度可变的蛋白质增强,增强了模型的泛化能力。模型在没有观察到的化合物或蛋白质情况下仍表现良好。

背景
化合物-蛋白质相互作用(CPI)预测旨在原图测化合物-蛋白质相互作用的模式和强度,以实现合理的药物发现。现有的基于深度学习的方法仅利用蛋白质序列或结构的单一模态,缺乏对两种模态联合分布的联合建模,这可能会导致复杂的现实场景中由于各种因素而导致性能显着下降。这些方法只在单一的固定尺度上模拟蛋白质序列和结构,而忽略了更细粒度的多尺度信息(例如嵌入在关键蛋白质片段中的信息)。
本研究提出了一种多尺度蛋白质序列-结构对比架构,通过模态内和跨模态对比来捕获蛋白质序列和结构的依赖关系。并进一步应用长度可变的蛋白质增强,以允许在不同尺度上的进行对比,从氨基酸等级到序列等级。
在训练过程中既没有观察到的化合物也没有观察到的蛋白质的情况下,模型都有很好的性能,此外在只有单模态的情况下也具有很好的性能。
CPI预测的理想框架通常应该是高效、有效和可推广的,而来自真实数据的两个主要瓶颈可能会阻碍CPI方法的发展。
**模态缺失:**序列-结构的联合建模对训练过程中的CPI预测有很大的好处,但在实际推理中经常遇到模态缺失的问题,即只有一种蛋白质模态,要么是序列,要么是结构,可以进行推理。更重要的是,我们无法预先假设我们可以获得哪种模式的蛋白质数据(或两者都有)。
**域转移:**大多数现有方法在训练集同源测试数据上工作良好,但很难推广到更实际的(训练集异源)测试数据,其中化合物,蛋白质或两者在训练期间从未观察到。
方法
化合物表示:一个化合物可以表示为一个分子图
G
C
=
(
ν
C
,
ξ
C
)
G_C=(\nu _C,\xi_C )
GC=(νC,ξC),其中每个节点
a
i
∈
ν
C
a_i \in \nu_C
ai∈νC化合物中的一个原子,每个边
e
i
,
j
∈
ξ
C
e_{i,j} \in \xi_C
ei,j∈ξC表示原子
a
i
a_i
ai和
a
j
a_j
aj之间的化学键。
蛋白质表示:含有
N
p
N_p
Np个氨基酸残基的蛋白质可以用序列
S
=
(
r
1
,
r
2
,
.
.
.
,
r
N
p
)
S=(r_1,r_2,...,r_{N_p})
S=(r1,r2,...,rNp)表示,蛋白质的氨基酸序列
S
S
S可以折叠成稳定的结构
G
p
G_p
Gp,形成一类特殊的多模态数据
P
=
(
S
,
G
p
)
P=(S,G_p)
P=(S,Gp)。其中蛋白质结构可以建模为蛋白质图
G
p
=
(
ν
p
,
ξ
p
)
G_p=(\nu_p,\xi_p)
Gp=(νp,ξp)。
给定
N
N
N个蛋白质
{
P
i
=
(
S
(
i
)
,
G
P
(
i
)
)
}
i
=
1
N
\left \{ P^{i}=(S^{(i)},G_{P}^{(i)}) \right \}_{i=1}^{N}
{Pi=(S(i),GP(i))}i=1N,
M
M
M个化合物
{
G
C
(
j
)
}
j
=
1
M
\left \{ G_{C}^{(j)} \right \} _{j=1}^{M}
{GC(j)}j=1M,化合物-蛋白质相互作用预测旨在预测化合物和蛋白质之间的相互作用模式
G
C
×
P
⟶
[
0
,
1
]
N
C
×
N
p
G_{C} \times P \longrightarrow [0,1]^{N_{C} \times N_{p}}
GC×P⟶[0,1]NC×Np和相互作用强度
G
C
×
P
⟶
R
≥
0
G_{C} \times P \longrightarrow R \ge 0
GC×P⟶R≥0
CPI预测的一般框架
1、给定的化合物中提取的化合物表示的化合物编码器
2、从给定蛋白质序列、结构或者两种模式中提取蛋白质表示的多模态蛋白质表示学习
3、成对相互作用模式预测,用于预测蛋白质残基与化合物原子之间的接触图,并学习蛋白质与化合物的联合表示
4、相互作用强度预测,用于预测化合物与蛋白质的结合亲和力
PSC-CPI架构图如图2所示:
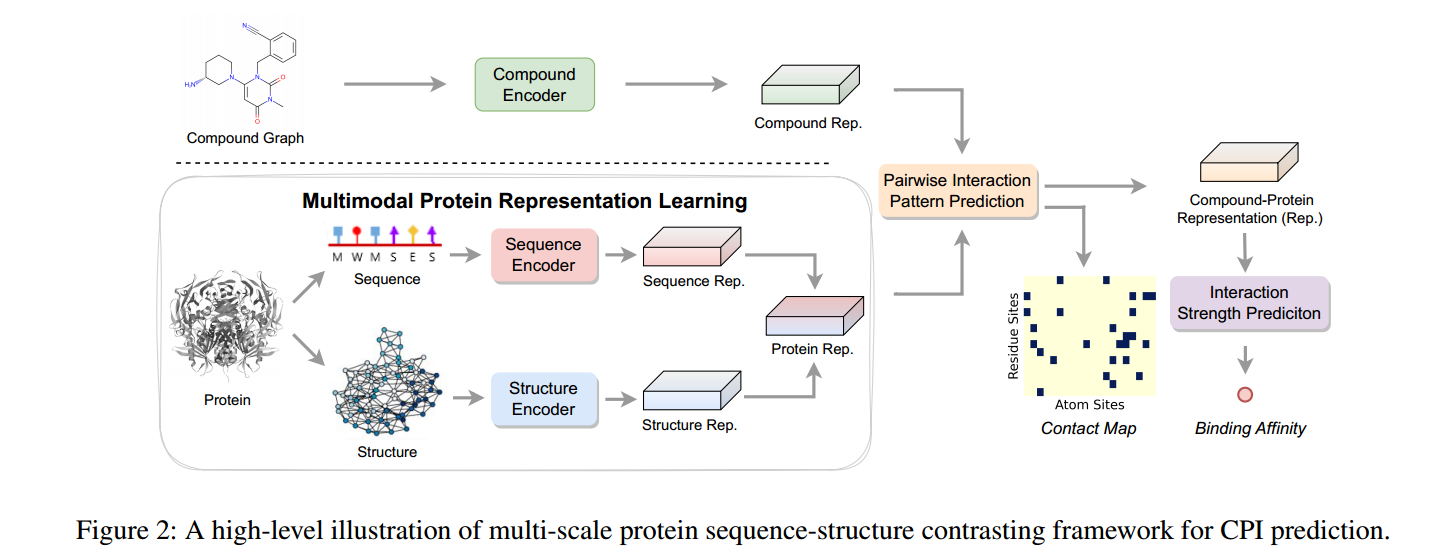
化合物编码器
化合物编码器以分子图
G
C
=
(
ν
C
,
ξ
C
)
G_C=(\nu _C,\xi_C )
GC=(νC,ξC)为输入,学习每个原子的
F
F
F维节点表示。
本文采用3层GCN作为编码器,给定一个分子图
G
C
=
(
ν
C
,
ξ
C
)
G_C=(\nu _C,\xi_C )
GC=(νC,ξC),GCNs将其邻接矩阵
A
c
A_c
Ac和节点特征
X
c
X_c
Xc作为每个节点的输入和输出表示。
Z
c
o
m
p
=
A
^
σ
(
A
^
σ
(
A
^
X
c
W
0
)
W
1
)
W
2
Z^{comp}=\hat{A} \sigma (\hat{A} \sigma(\hat{A} X_c W^0)W^1)W^2
Zcomp=A^σ(A^σ(A^XcW0)W1)W2
其中
σ
\sigma
σ为激活函数
R
E
L
U
(
∗
)
RELU(*)
RELU(∗),
A
^
=
D
^
1
/
2
(
A
c
+
I
)
D
^
−
1
/
2
\hat{A}=\hat{D}^{1/2}(A_c+I) \hat{D}^{-1/2}
A^=D^1/2(Ac+I)D^−1/2表示标准化邻接矩阵,
I
I
I为单位矩阵,
D
^
\hat{D}
D^为
(
A
c
+
I
)
(A_c+I)
(Ac+I)的对角度矩阵。
成对相互作用模式预测
该模块,以化合物表示
Z
c
o
m
p
Z^{comp}
Zcomp和蛋白质表示
Z
p
r
o
t
Z^{prot}
Zprot作为输入,首先通过独立的线性变换
W
c
o
m
p
和
W
p
r
o
t
W^{comp}和W^{prot}
Wcomp和Wprot,将他们转换成一个低维潜在空间,然后通过内积计算每个残基-原子对的相互作用强度(intensity),并进行归一化。得到第m个原子和第n个残基的相互作用强度
P
c
o
n
t
[
m
,
n
]
P^{cont}[m,n]
Pcont[m,n]。
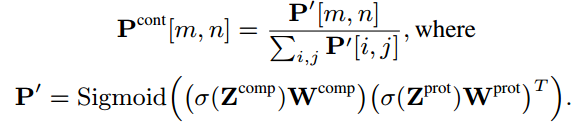
为了获得化合物-蛋白质联合嵌入,我们计算了每个残基-原子对表示的Hadamard乘积,并将它们与
P
c
o
n
t
P^{cont}
Pcont作为权重相加。
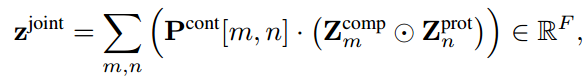
相互作用强度(Strength)预测
将他们的联合嵌入
z
j
o
i
n
t
z^{joint}
zjoint作为输入,并将其从高维空间映射到一个非负值
y
a
f
f
y^{aff}
yaff,即亲和力。该模块由一个1D卷积层Conv(·)、一个最大池化层Max(·)和一个3层多层感知器MLP(·)组成,表示为:

多尺度序列-结构对比
本文提出了一个多尺度序列结构对比框架,如图3所示,该框架通过长度可变的蛋白质扩增和内/跨模态对比,充分捕获了序列结构依赖关系和多尺度信息。

长度可变蛋白质扩增
数据增强的主要目的是生成与原始视图共享相同或相似语义的不同增强视图。
蛋白质数据增强的两个主要挑战:
1、长度的多样性,不同蛋白质可能具有不同的序列长度
2、关键片段的变异性,不同蛋白质上的关键片段可能具有非常不同的长度并且位于序列上的不同位置
为了应对这些挑战,我们通过从整个蛋白质序列中采样长度可变的连续片段(子序列)并提取相应的子图来增强蛋白质数据。传统的增强方法,通常采样相同长度或者长度比的蛋白质子序列,在训练之前对其进行修复。
本文中,为每个蛋白质
P
=
(
S
(
i
)
,
G
p
(
i
)
)
P=(S^{(i)},G_p^{(i)})
P=(S(i),Gp(i))在每个训练epoch, 生成不同长度的增强子序列
{
S
(
i
,
j
)
}
k
=
1
K
\left \{S^{(i,j)}\right \}_{k=1}^{K}
{S(i,j)}k=1K和相应子图
{
G
p
(
i
,
j
)
}
k
=
1
K
\left \{G_p^{(i,j)}\right \}_{k=1}^{K}
{Gp(i,j)}k=1K
随着训练的进行,增强的蛋白质子序列的长度不断变化,使的该模型能够在更多不同的尺度上“看到”相同的蛋白质,从而捕获蛋白质中更多的多尺度信息。
模态内和跨模态对比
我们的框架中引入了两种不同的对比学习目标,即模态内对比和跨模态对比,以捕获蛋白质序列或结构以及跨模态依赖性内的多尺度信息。
首先将第i个蛋白质序列
S
(
i
)
S^{(i)}
S(i)和增强子序列
{
S
(
i
,
j
)
}
k
=
1
K
\left \{S^{(i,j)}\right \}_{k=1}^{K}
{S(i,j)}k=1K输入到序列编码器
f
s
e
q
(
.
)
f_{seq}(.)
fseq(.),输出序列表示如下:
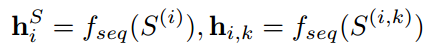
同理结构编码器的输出结构表示:
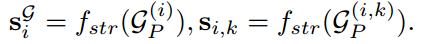
更随 SimCLR (Chen et al 2020b) 和 JOAO (You et al 2021) ,两个不同的两层 MLP 投影头,表示为 g1(·) 和 g2(·),进一步应用于将序列和结构表示映射到低维空间。
接下来,定义由模态内对比和跨模态对比组成的对比目标函数来预训练序列和结构编码器。对于模态内对比,我们将蛋白质序列(蛋白质图)及其子序列(子图)视为正对,将同一批次中其他蛋白质(蛋白质图)的子序列视为负对,可以定义为:
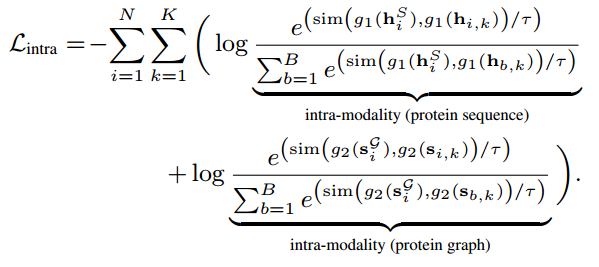
B为批次,sim(.,.)为余弦相似度,τ是温度系数,模态内对比从通过最大化子序列(子图)和全序列(蛋白质图)的互信息,将不同长度的蛋白质片段转化为最终表示迁移知识。
跨模态对比旨在通过使同一蛋白质片段的子序列和子图在不同尺度上共享相似的语义来捕获序列结构依赖性。具体来说,跨模态对比将来自同一蛋白质的子序列和子图视为正对。
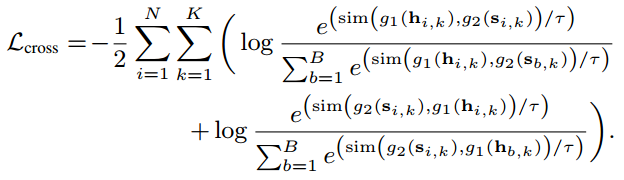
最后,用于预训练序列和结构编码器的总损失函数可以定义为:
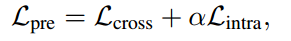
Train and Inference
Train
CPI预测主要有两个下游任务,即强度预测和模式预测
当我们知道第i个蛋白质和第j个化合物之间真实的相互作用强度
y
i
,
j
t
r
u
e
y_{i,j}^{true}
yi,jtrue,CPI强度预测的目标函数定义如下:
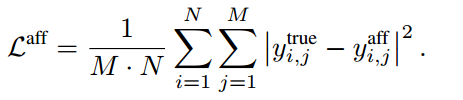
类似地,如果我们知道第 i 个蛋白质和第 j 个化合物之间的真实相互作用模式
P
i
,
j
t
r
u
e
P_{i,j}^{true}
Pi,jtrue,则 CPI 模式预测的目标函数定义如下,
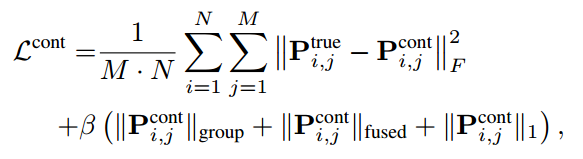
其中
∥
P
i
,
j
c
o
n
t
∥
g
r
o
u
p
\left \| P_{i,j}^{cont} \right \|_{group}
Pi,jcont
group、
∥
P
i
,
j
c
o
n
t
∥
f
u
s
e
d
\left \| P_{i,j}^{cont} \right \|_{fused}
Pi,jcont
fused和
∥
P
i
,
j
c
o
n
t
∥
1
\left \| P_{i,j}^{cont} \right \|_{1}
Pi,jcont
1是是 ( Explainable deep relational networks for predicting compound–protein affinities and contacts.) 采用的三种结构感知稀疏正则化控制交互接触图


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









