







| 题目 | Contrastive learning in protein language space predicts interactions between drugs and protein targets |
|---|---|
| MIT | |
| 期刊/年份 | PNAS/2023 |
| 状态 | 阅读中 |
| DOI | https://doi.org/10.1073/pnas.2220778120 |
| Github | https://github.com/samsledje/ConPLex |
摘要
基于序列的药物-靶标相互作用预测有可能通过补充实验筛选来加速药物发现。这种计算预测需要具有通用性和可扩展性,同时对输入中的细微变化保持敏感。然而,目前的计算技术不能同时满足这些目标,往往牺牲一个性能来实现另一个。本研究提出了一个利用预训练的蛋白质语言模型(“Plex”)结合蛋白质锚定对比嵌入(“Con”)的深度学习模型-ConPlex,实现了高精度以及对未见数据的适应性和对诱饵化合物的特异性。它根据学习到的表示之间的距离进行结合预测,从而能够在大规模化合物库和人类蛋白质组规模上进行预测。此外模型嵌入具有可解释性,能够可视化药物靶标嵌入空间并使用嵌入来表征人类细胞表面的蛋白功能。
背景意义
在药物发现流程中,主要限制是针对感兴趣的蛋白质靶点对潜在的药物分子进行实验筛选。因此快速准确的计算预测药物-靶标相互作用很有价值。
分子对接是一类重要的计算DTI方法,它使用药物和靶标的三维结构表示。虽然最近高通量精确的3D蛋白质结构预测模型的可用性意味着这些方法可以仅从蛋白质的氨基酸序列开始使用,但遗憾的是,分子对接和其他基于结构的方法(例如,合理设计,活性位点建模,模板建模)的计算费用对于大规模DTI筛选仍然令人难以接受。
另一类DTI预测方法仅隐式地使用3D结构,当输入仅包含药物的分子描述[如SMILES字符串]和蛋白质靶标的氨基酸序列时,可以进行快速的DTI预测。这类基于序列的DTI方法能够实现可扩展的DTI预测,但是在对比基于结构的方法获得的精度水平方面存在障碍。
模型(ConPlex)
本文介绍了一个基于序列的DTI预测方法(ConPlex)。它利用预训练的蛋白质语言模型(PLMs)的丰富特征,并表明他可以在大规模DTI预测任务中产生先进的预测性能。ConPlex中的基于信息的PLM和对比学习克服了以往方法的局限性。虽然已经提出了许多方法用于DTI问题的基于序列的方法[例如,使用联邦学习,CNN, Transformer],但它们的蛋白质和药物表示仅由DTI基础数据构建。DTI输入之间的高度多样性,加上DTI训练数据的有限可用性,限制了这些方法的准确性及其在训练域之外的推广能力。此外,确实一般方法往往是通过牺牲细粒度的特异性来实现的,即无法区分具有相似物理化学性质的真阳性结合化合物和假阳性结合化合物(诱饵,Decoy)。
相比之下,ConPLex的“PLex”(预训练词典)部分有助于缓解DTI训练数据有限的问题。解决DTI数据集有限的问题的一种方法是将学习到的蛋白质表示从预训练的plm转移到DTI预测任务中。(PLMs可以有助于解决DTI数据集有限的问题)。DTI的数据集的大小限制了以前方法学习到的表示质量。PLMs以无监督的方式学习数百万蛋白质中氨基酸序列的分布特征,生成基于序列的表示。机器学习中的一个设计范例是,输入的信息特征可以增强模型的能力。我们通过使用“Con”(对比学习)部分直接解决了我们架构中的细粒度特异性问题:一种蛋白质锚定的对比共嵌入,将蛋白质和药物放置在共享的潜在空间中。我们表明,这种共嵌入将真正的相互作用和诱饵强制分离,以实现广泛的泛化和高特异性。ConPlex泛化能力强,及时训练样本不含有给定测试样本信息的情况下也能实现较好的结果(zero-shot)。
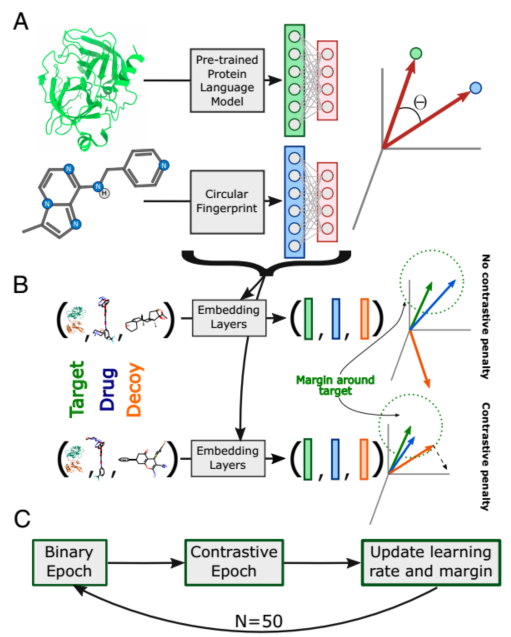
图2
A:蛋白质特征使用预训练的PLM(ProtBert)生成,药物特征使用Morgan fingerprint生成,这些特征通过学习到的非线性投影转化到共享的潜在空间。相互作用的预测基于该空间中的余弦距离,转换参数通过低覆盖率数据集上的二元交叉熵(BCE)进行更新。
B:在对比阶段,靶标、药物和Decoy的三联体以同样的方式转化到共享空间。转换被视为一个度量学习问题。使用高覆盖数据集(36)上的三元组距离损失来更新参数,以最小化目标-药物距离,同时最大化目标-诱饵距离。如果目标与诱饵之间的距离大于目标与药物之间的距离加上一些余量,则不施加额外的处罚。
C:ConPLex 在A阶段和对比阶段交替进行训练,以同时优化两个目标。每一轮训练后,学习率和对比边际都会根据退火方案进行更新。
ConPLex 比其他同类方法能更准确地预测 DTI。并且结合迁移学习,模型可针对特定任务调整基础模型。特别是,我们发现现有的基于序列的 DTI 预测方法对数据集中药物与蛋白质覆盖率的变化非常敏感,而 ConPLex 在多种覆盖率情况下都表现出色。ConPLex 在zero-shot中表现尤为出色(在训练时没有关于给定蛋白质或药物的信息)。通过实验验证,ConPLex 的命中率为 63%(12/19),其中包括 4 个亚纳摩尔结合亲和力的命中,这证明了 ConPLex 作为一种精确、可高度扩展的硅学筛选工具的价值。
此外ConPlex不仅可以预测相互作用还可以预测结合亲和力,共享空间除了具有预测准确性还提供了可解释性,文中证明了该空间中的距离能有意义地反映蛋白质结构域和结合功能(利用 ConPLex 表示法从功能上描述了 Surfaceome 数据库中的细胞表面蛋白,这是一组定位于外部质膜的 2,886 个蛋白,它们参与信号传导,很可能容易被配体靶向).
ConPLex非常快:作为概念验证,使用单个NVIDIA A100 GPU在不到24小时内对ChEMBL 中所有药物-人类蛋白质组进行预测大约有
2
∗
1
0
10
2*10^{10}
2∗1010。因此,ConPLex有潜力应用于需要大量计算的任务。注意到大多数DTI方法需要对每个药物-靶标对进行大量计算(即具有二次时间复杂度)。由于ConPLex预测仅依赖于联合空间中的距离,因此只要计算并对齐embedding(具有线性时间复杂度),就可以高效地进行预测。
区分低覆盖率和高覆盖率 DTI 预测
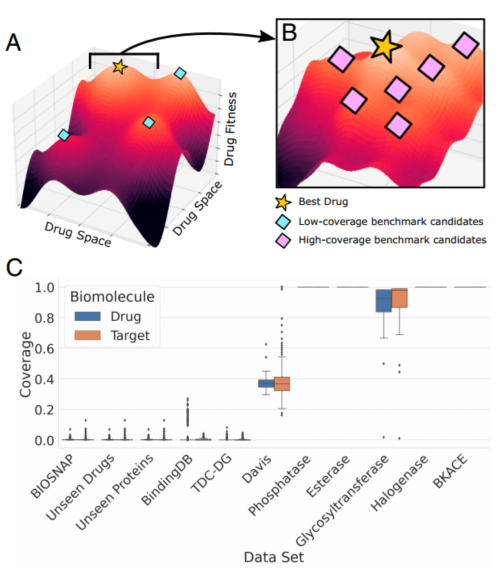
图1药物-靶点相互作用基准的覆盖率差异很大。覆盖率被定义为数据集中存在数据点(阳性或阴性)的药物或靶点的比例。高覆盖率与低覆盖率基准往往会奖励不同类型的模型性能。
(A) 在这个低覆盖率数据集示例漫画中,候选药物覆盖了整个空间的多样性,没有两种药物高度相似。一个成功的模型可以学习到对适合度景观的粗略估计,但必须对药物空间的大部分进行精确建模,才能推广到所有候选药物。
(B) 对于高覆盖率数据集,药物往往针对特定的蛋白质家族。因此,一个成功的模型不需要概括如此广泛的范围,但必须能够捕捉药物适应性中更微小的变化,以实现高特异性并区分类似药物。
(C)对现有流行的DTI基准数据集的回顾中,现有基准覆盖范围差异很大,从几乎为零覆盖的数据集(每种药物或靶标只出现几次)到几乎完全覆盖的数据集(所有药物或靶标在数据中都是已知的)。
ConPLex在这两种情况下都优于其竞争对手,但请注意,将两种机制分开有助于澄清该领域一个经常看到的问题:在不同提DTI基准中,其性能差异很大。之前已经进行了几次尝试,以使DTI基准标准化,并为模型评估开发一致的框架。然而,这项工作的大部分都忽略了基准测试的一个关键—不同的生物分子数据覆盖率,我们发现这个方面会显著影响模型性能差异。我们将覆盖率定义为该数据集中存在数据点的药物或靶点的平均比例,无论这是积极的还是消极的相互作用(方法)。根据基准数据集的每生物分子数据覆盖率,我们声称这些基准研究的是非常不同的问题。
低覆盖率(图1A)倾向于测量DTI领域的广泛范围,包含高度多样化的药物和靶点。由于所覆盖目标的多样性,这些数据集可能会带来建模挑战,但允许对化合物和蛋白质类别之间的兼容性进行广泛评估。
当将DTI模型应用于大规模的扫描以预测潜在的对于大型化合物文库的相互作用时,就和低覆盖率机制相关。每个独特的生物分子只有少量已知的相互作用。因此,重要的是用于这些任务的DTI模型具有广泛的适用性,并且可以准确地推广到许多不同的蛋白质和药物家族。然而,这种泛化往往以特异性为代价,导致模型无法区分高度相似的药物或蛋白质。
高覆盖率数据集(图1B)代表了相反的权衡:它们在药物或靶标类型上包含有限的多样性,但具有密集的潜在两两相互作用。因此,它们捕获了药物靶标结合的特定亚类的细粒度细节,并能够在特定环境中区分相似的生物分子。
当优化特定的交互时,高覆盖率是相关的。在这里,可以训练模型对蛋白质家族或药物类别具有高度特异性,以至于训练每个药物或每个靶标模型来捕获该生物分子的精确结合动力学。这种模型缺乏泛化超出训练领域的能力,因此不能用于基因组或药物库规模的预测。
ConPLex的PLM方法在两种情况下都具有强大的性能。在低覆盖率情况下,优势主要来自“PLex”部分,它可以利用语言模型的有效泛化来实现最先进的性能。在高覆盖率的数据集上,“Con”部分(对比学习)也变得很重要,因为以高精度训练特定药物或目标的模型变得可行,而且这些模型通常比更通用的模型表现得更好。虽然单任务模型在给定可用数据的情况下确实表现良好,但ConPLex能够在低多样性、高覆盖率的情况下实现极高的特异性,同时仍然广泛适用于数据有限的蛋白质靶点。因此,ConPLex既适用于大规模的化合物或目标筛选,也适用于细粒度、高度特异性的结合预测。
Methods
计算数据覆盖率
1
(
i
,
j
)
1_{(i,j)}
1(i,j)为指标变量,表示药物i和靶点j的观测值。
对于一个有m个唯一药物和n个唯一靶点的数据集,我们可以定义药物d的覆盖率为
C
d
=
1
n
∑
j
=
0
n
1
(
d
,
j
)
C_d=\frac{1}{n} {\textstyle \sum_{j=0}^{n}} 1_{(d,j)}
Cd=n1∑j=0n1(d,j),可以定义靶点t的覆盖率为
C
t
=
1
m
∑
i
=
0
m
1
(
i
,
t
)
C_t=\frac{1}{m} {\textstyle \sum_{i=0}^{m}} 1_{(i,t)}
Ct=m1∑i=0m1(i,t)。然后,对于给定的数据集,我们可以评估drug和target覆盖度中值。
一个数据集如果具有最大覆盖度,那么对于每个药物-靶标对(即每个特定的药物与靶标的组合),将会有一个单独的数据点。这也意味着数据集中包含了对每种药物和每个靶标的详细信息,没有重复的数据。
这个观点通常用于描述数据集的信息覆盖程度。在这个情境中,“coverage” 表示数据集中包含的不同药物-靶标对的数量。如果每种药物和每个靶标都有大量的数据点,那么覆盖度就很高。而如果数据集中只包含每种药物和每个靶标的一个数据点,那么覆盖度就很低,因为它没有提供有关药物和靶标的多样性信息。
如果数据集的药物空间和靶标空间都很大,并且都全连接,就能包含泛化信息(低覆盖)和特异性信息(高覆盖)。然而,通常是没有这种数据集的,高覆盖数据集一般总是不包含足够的多样性来训练模型广泛应用于新的药物或目标。
数据集介绍
低覆盖率benchmark
作者在三个大规模、低覆盖率的基准数据集上评估模型,DAVIS和BindingDB由实验确定的解离常数( K D K_D KD)的药物和靶点对组成,根据文献_(MolTrans: Molecular interaction transformer for drug–target interaction prediction)_,作者将 K D < 30 K_D<30 KD<30视为正DTI,将 K D > = 30 K_D>=30 KD>=30视为负DTI,第三个数据集,来自BIOSNAP的ChGMiner,仅由正DTI组成。我们通过随机取样等量的蛋白质-药物对来产生负DTI,并假设随机的一对不太可能产生正相互作用。DAVIS数据集代表了 few-shot learning setting:它只包含2,086个训练互作,而BindingDB和BIOSNAP分别有12,668个和19,238个。这些数据集被分成70%用于训练,10%用于验证,剩下的20%用于测试。训练数据被进行下采样,使其具有相同数量的正相互作用和负相互作用,而验证和测试数据则保留在数据集中原始的比例。
zero-shot benchmarks
作者用BIOSNAP的两种zero-shot修改来评估方法。通过从全套蛋白质中选择20%的蛋白质并选择包括这些蛋白质的任何相互作用作为测试集,创建了Unseen蛋白质集。因此,没有蛋白质同时出现在训练集和测试集。使用相应的过程来创建Unseen药物集。然后进一步划分训练集,使用7/8的交互作用进行训练,1/8的交互作用进行验证。如上所述,对数据进行人为二次采样,以便平衡训练。
连续benchmark
连续的亲和力预测数据来自TDC-DG(Therapeutics data commons: Machine learning datasets and tasks for drug discovery and development)。TDC-DG由140,746种唯一药物和477种唯一靶点组成,这些靶点来自BindingDB的相互作用。每个相互作用都用实验确定的解离常数 I C 50 IC_{50} IC50来标记。使用由TDC基准测试框架确定的训练与验证分割来训练模型,并报告测试集中预测的平均PCC系数。
高覆盖benchmark
来自Useful Decoys数据库:增强的DUD-E(Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking)由102个蛋白质靶点和已知的结合伙伴(平均每个靶点224个分子)组成。对于每个结合伙伴,有50个decoys,或物理化学上类似的化合物,已知不会与靶标结合。值得注意的是,其中57个靶标被分类为GPCR、激酶、核蛋白或蛋白酶。通过在类内分割靶点来生成训练-测试分割,这样在训练集和测试集中都有每个类的代表性成员。根据定义,这些数据具有高覆盖率,因为每个靶点都有几种真实化合物和诱饵化合物。
ConPlex模型
靶标特征
使用预训练的PLMs生成蛋白质目标特征,对于一个长度为n的蛋白质得到嵌入矩阵为:
E
f
u
l
l
∈
R
n
×
d
t
E_{full} \in R^{n\times d_t}
Efull∈Rn×dt
如果沿着蛋白质长度进行平均池化,可得到向量:
E
∈
R
d
t
E \in R^{d_t}
E∈Rdt
作者研究了,预训练模型Prose、esm、protBert,其默认维度分别为
d
t
d_{t}
dt = 6165、1280和1024 (SI附录,S2)。Elnaggar等人推荐使用ProtT5XLUniref50,但是我们发现它在DTI预测任务上的表现不如ProtBert。
药物特征
作者通过药物分子的摩根指纹(The generation of a unique machine description for chemical structures—A technique developed at chemical abstracts service)来表征药物分子,这是分子图的SMILES字符串的编码,通过考虑每个原子周围的局部邻域,将其作为固定维嵌入 M ∈ R d m M \in R^{d_m} M∈Rdm,选择 d m = 2048 d_{m}=2048 dm=2048。摩根指纹对小分子表征的效用已在很多实验中得到证明。
转化到共享潜在空间
给定靶标嵌入
T
∈
R
d
t
T \in R^{d_t}
T∈Rdt和小分子嵌入
M
∈
R
d
m
M \in R^{d_m}
M∈Rdm,通过一个具有Relu激活函数的单全连接层将它们转换为
T
∗
,
M
∗
∈
R
h
T^*,M^* \in R^h
T∗,M∗∈Rh,权重矩阵为
W
t
∈
R
h
×
d
t
,
W
m
∈
R
h
×
d
m
W_t \in R^{h\times d_t},W_m \in R^{h\times d_m}
Wt∈Rh×dt,Wm∈Rh×dm,偏置向量为
b
t
,
b
m
∈
R
h
b_t,b_m \in R^h
bt,bm∈Rh.
T
∗
=
R
e
l
u
(
W
t
T
+
b
t
)
T^*=Relu(W_tT+b_t)
T∗=Relu(WtT+bt)
M
∗
=
R
e
l
u
(
W
m
T
+
b
m
)
M^*=Relu(W_mT+b_m)
M∗=Relu(WmT+bm)
给定潜在的嵌入
T
∗
,
M
∗
T^*,M^*
T∗,M∗,通过计算两个嵌入向量之间的余弦相似度来作为一个DTI的概率
P
^
(
T
∗
,
M
∗
)
\hat{P} (T^*,M^*)
P^(T∗,M∗),然后使用sigmoid激活函数,因此我们将预测概率定义为:
p
^
(
T
∗
,
M
∗
)
=
σ
(
T
∗
⋅
M
∗
∥
T
∗
∥
2
⋅
∥
M
∗
∥
2
)
\hat{p}(T^*,M^*)=\sigma (\frac{T^*\cdot M^*}{\left \| T^* \right \|_2 \cdot \left \| M^* \right \|_2 } )
p^(T∗,M∗)=σ(∥T∗∥2⋅∥M∗∥2T∗⋅M∗)
当预测靶标与药物亲和力
y
^
(
T
∗
,
M
∗
)
\hat{y} (T^*,M^*)
y^(T∗,M∗),
y
^
(
T
∗
,
M
∗
)
=
R
e
l
u
(
T
∗
⋅
M
∗
)
\hat{y} (T^*,M^*)=Relu(T^* \cdot M^*)
y^(T∗,M∗)=Relu(T∗⋅M∗)
Train
对模型进行广义和精细预测的训练,并根据训练数据集计算损失。大范围训练数据(低覆盖率)在真实标签y和预测标签之间使用二元交叉熵损失(LBCE),当模型被训练来预测结合亲和力时,我们在监督过程中用均方误差损失(LMSE)代替二元交叉熵损失。
使用对比学习在精细化数据训练(高覆盖率数据),对比学习使用三组训练点而不是成对,分别表示锚点、正点和负点,目的是最小化锚点与正例之间的距离,而最大化锚点与负例之间的距离。在DTI设置中,三联体的自然选择分别是蛋白质靶点作为锚点,真正的药物作为阳性,诱饵作为阴性。
对于每个已知的相互作用的药物-靶标对(T, M+),我们随机抽取k = 50个非相互作用的药物-靶标对(T, M−),并生成三联体(T, M+, M−),其中
M
−
M^-
M−是从针对T的所有诱饵集合中抽取的。我们可以计算三重边缘距离损失(
L
T
R
M
L_{TRM}
LTRM)
L
T
R
M
(
a
,
p
,
n
)
=
1
n
∑
i
=
1
N
m
a
x
(
D
(
a
,
p
)
−
D
(
a
,
n
)
+
m
,
0
)
L_{TRM}(a,p,n)=\frac{1}{n}\sum_{i=1}^{N}max(D(a,p)-D(a,n)+m,0)
LTRM(a,p,n)=n1∑i=1Nmax(D(a,p)−D(a,n)+m,0)
D
(
u
,
v
)
=
1
−
p
^
(
u
,
v
)
D(u,v)=1-\hat{p}(u,v)
D(u,v)=1−p^(u,v)
m
m
m表示边际增量,此处
m
=
0
m=0
m=0
Margin annealing(边际退火)
最初,较大的差额要求诱饵比药物离目标远得多,以避免惩罚,从而导致更大的权重更新。随着训练的进行,较低的边际放宽了这一限制,只要求药物比诱饵更接近
m
⟶
0
m\longrightarrow 0
m⟶0,根据一个tanh衰减与重启调度,margin初始设置为
M
m
a
x
=
0.25
M_{max}=0.25
Mmax=0.25.
每次
E
m
a
x
=
10
E_{max}=10
Emax=10个对比epoch,边界被重置为初始的
M
m
a
x
M_{max}
Mmax,总共50个epoch。在epoch i,边距设置为:
m
(
i
)
=
M
m
a
x
(
1
−
t
a
n
h
(
2
(
i
m
o
d
E
m
a
x
)
E
m
a
x
)
)
m(i)=M_{max}(1-tanh(\frac{2(i\ mod\ E_{max})}{E_{max}} ))
m(i)=Mmax(1−tanh(Emax2(i mod Emax)))

















 569
569

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









