







参考链接:
https://www.bioconductor.org/packages/devel/workflows/vignettes/RNAseq123/inst/doc/limmaWorkflow_CHN.html
https://cloud.tencent.com/developer/article/1422739
1. 加载包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("airway", quietly = TRUE)) #使用数据集
BiocManager::install("airway")
if (!require("org.Hs.eg.db", quietly = TRUE)) #注释基因
BiocManager::install("org.Hs.eg.db")
if (!require("clusterProfiler", quietly = TRUE)) #ID转换
BiocManager::install("clusterProfiler")
if (!require("edgeR", quietly = TRUE)) #过滤低表达基因
BiocManager::install("edgeR")
2. 导入数据
options(stringsAsFactors = F)
library(airway)
data(airway)
# 获取基因counts矩阵
expr <- assay(airway)
expr[1:5,1:5]

#获取分组信息
group_list <- colData(airway)$dex
group_list

3. 注释基因
参考链接:http://www.360doc.com/content/19/0506/00/30846661_833639624.shtml
#查看支持的ID转换类型
keytypes(org.Hs.eg.db)

#转换ID
geneid<-bitr(rownames(expr), fromType='ENSEMBL', toType='SYMBOL', OrgDb='org.Hs.eg.db', drop = TRUE) #去除空值
geneid
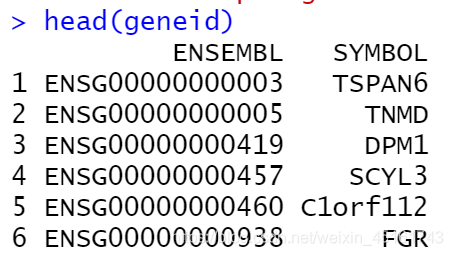
#注释ID
expr = expr[ rownames(expr) %in% geneid[ , 1 ], ]
geneid = geneid[ match(rownames(expr), geneid[ , 1 ] ), ]
rownames(expr)<-geneid$SYMBOL
expr[1:5,1:5]

- 常用ID
1)Ensemble id: 欧洲生物信息数据库提供,一般以ENSG开头,后边跟11位数字。如TP53基因:ENSG00000141510
2)Entrez id: 美国NCBI提供,通常为纯数字。如TP53基因:7157
3)Symbol id: 文献中报道的基因名称。如TP53基因的symbol id为TP53
4)Refseq id: NCBI提供的参考序列数据库:可以是NG、NM、NP开头,分别代表基因,转录本和蛋白质。如TP53基因的某个转录本信息可为NM_000546 - 基因组注释包: http://bioconductor.org/packages/release/BiocViews.html
4. 去重
table(duplicated(rownames(expr))) #统计重复基因数目
#对重复基因名取平均表达量
if(sum(duplicated(rownames(expr)))>0) #判断是否有重复基因
expr<-avereps(expr,ID=rownames(expr))

- 去重前:
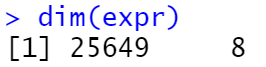
- 去重后:
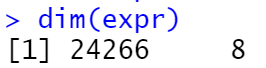
5. 过滤低表达基因
- 查看样本表达基因数目
apply(expr,2,fivenum)
apply(expr,2, function(x) sum(x>1))
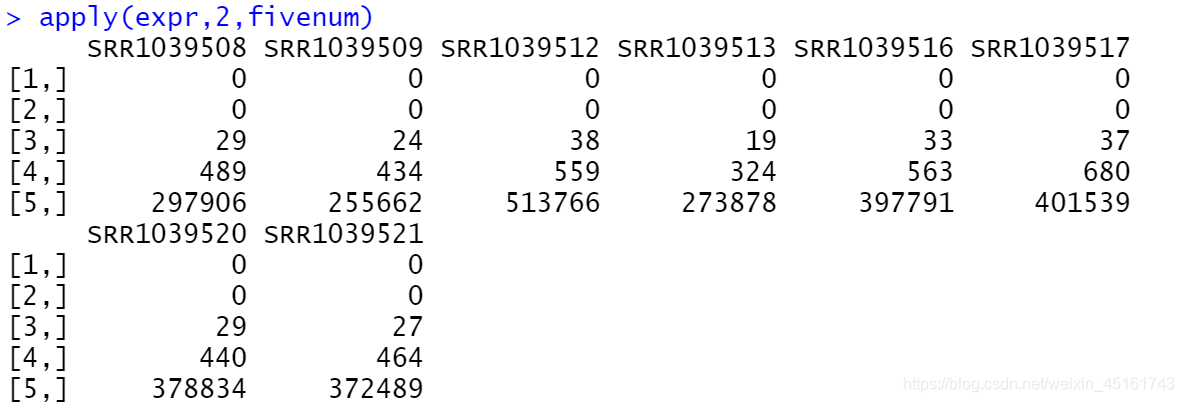
从五个分位数统计量可以发现,每个样本含有部分表达值为0的基因,需要过滤

从上图可以发现平均每个样本被检测到的基因数量是15600左右
- 过滤方法1
过滤所选用的阈值具有主观性
#过滤在所有样本都是零表达的基因
expr=expr[apply(expr,1, function(x) sum(x>1) > 1),]
dim(expr)
apply(expr,2,fivenum)

过滤了31%的低表达基因

- 过滤方法2
x <-edgeR::DGEList(counts=expr) #构建DEGList对象
keep.exprs <- edgeR::filterByExpr(x, group=group_list)#判断基因是否低表达
x <- x[keep.exprs,, keep.lib.sizes=FALSE]#过滤
head(keep.exprs)
dim(x)

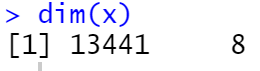
ps:
1)默认选取最小的组内的样本数量为最小样本数,保留至少在这个数量的样本中有10个或更多序列片段计数的基因
2)对基因表达量进行过滤时使用CPM值而不是表达计数,以避免对总序列数大的样本的偏向性
- 可视化
使用的是过滤方法2得到的数据
#得到数据
library(RColorBrewer)
x <-edgeR::DGEList(counts=expr,group=factor(group_list))#构建DGE对象
L <- mean(x$samples$lib.size)/10^6
M <- median(x$samples$lib.size)/10^6
lcpm <- cpm(x, log=TRUE, prior.count=2)#log2(CPM + 2/L)
lcpm.cutoff <- log2(10/M + 2/L) #过滤阈值
nsamples <- ncol(x) #提取样本数
col <- brewer.pal(nsamples, "Paired") #颜色
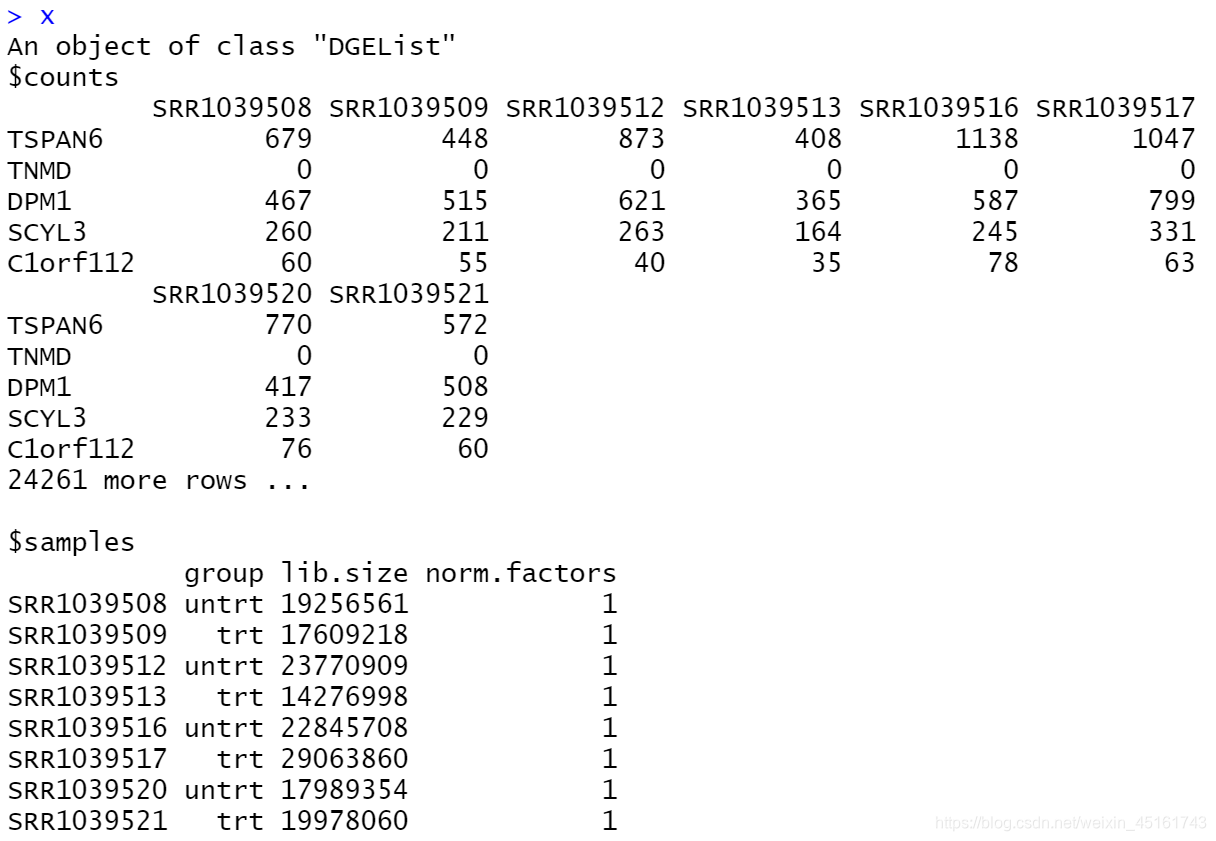
lib.size为各个样本的总read counts
par(mfrow=c(1,2)) #一式两图
#过滤前图形
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.26), las=2, main="", xlab="")
abline(v=lcpm.cutoff, lty=3) #阈值线
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
title(main="A. Raw data", xlab="Log-cpm") #标题和x轴名称
legend("topright",colnames(x), text.col=col, bty="n",cex=0.5) #图例
#过滤后图形
keep.exprs <- edgeR::filterByExpr(x, group=group_list)#判断基因是否低表达
x <- x[keep.exprs,, keep.lib.sizes=FALSE]#过滤
lcpm <- cpm(x, log=TRUE, prior.count=2)
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.26), las=2, main="", xlab="")
title(main="B. Filtered data", xlab="Log-cpm")
abline(v=lcpm.cutoff, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright",colnames(x), text.col=col, bty="n",cex=0.5)
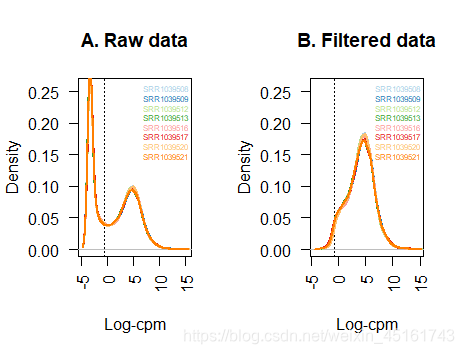
注: 卡阈值方法
6. 无监督聚类MDS图
参考链接:https://blog.csdn.net/yanta0/article/details/84798062
- 基本思想: 将高维坐标中的点投影到低维空间中,保持点彼此之间的相似性尽可能不变,而点与点之间的距离和对象之间的相似度高度相关
- 目的:
1)展示出样品间的相似性和不相似性
2)定性检测差异表达基因数目,越相似则差异基因越少
3)检查有无分组错误情况出现 - 理想情况: 样本会在不同的实验组内很好的聚类,且可以鉴别出远离所属组的样本
par(mfrow=c(1,1))
lcpm <- cpm(x, log=TRUE)
col.group <- group_list
levels(col.group) <- brewer.pal(nlevels(col.group), "Set1")
col.group<-droplevels(col.group) #去除不使用的水平因子
col.group <- as.character(col.group)
plotMDS(lcpm, labels=group, col=col.group)
title(main="Sample groups")


7. 保存数据
保留的是使用过滤方法1得到的数据
save(group_list,expr,file="expr.RData")



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









