







参考链接:http://blog.sciencenet.cn/blog-3406804-1188480.html
#加载包和数据
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("edgeR", quietly = TRUE))
BiocManager::install("edgeR")
options(stringsAsFactors = F)
load("expr.RData")
expr.RData储存了经过处理后的表达矩阵和分组信息,详见差异分析流程(一)数据预处理

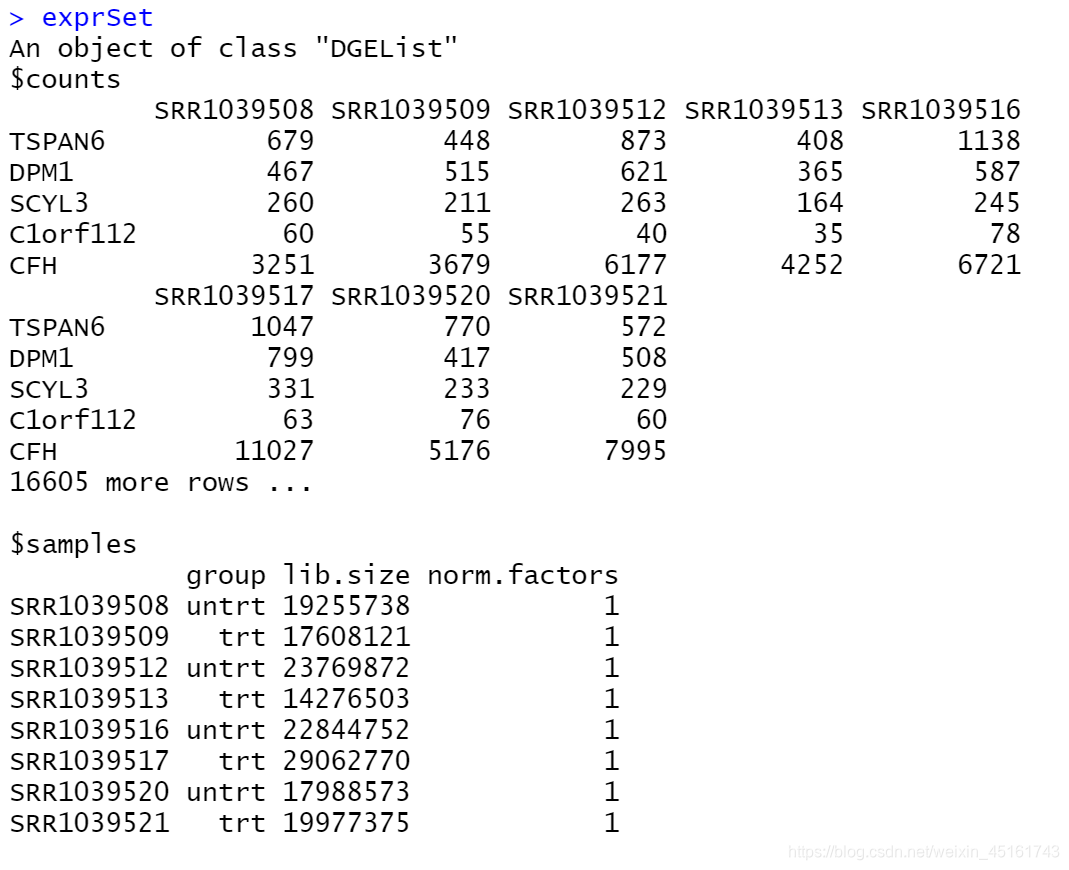
1.构建DGEList对象
- 输入: raw counts或RSEM的counts,不推荐输入cufflinks得到的counts
exprSet <- DGEList(counts = expr, group = group_list)
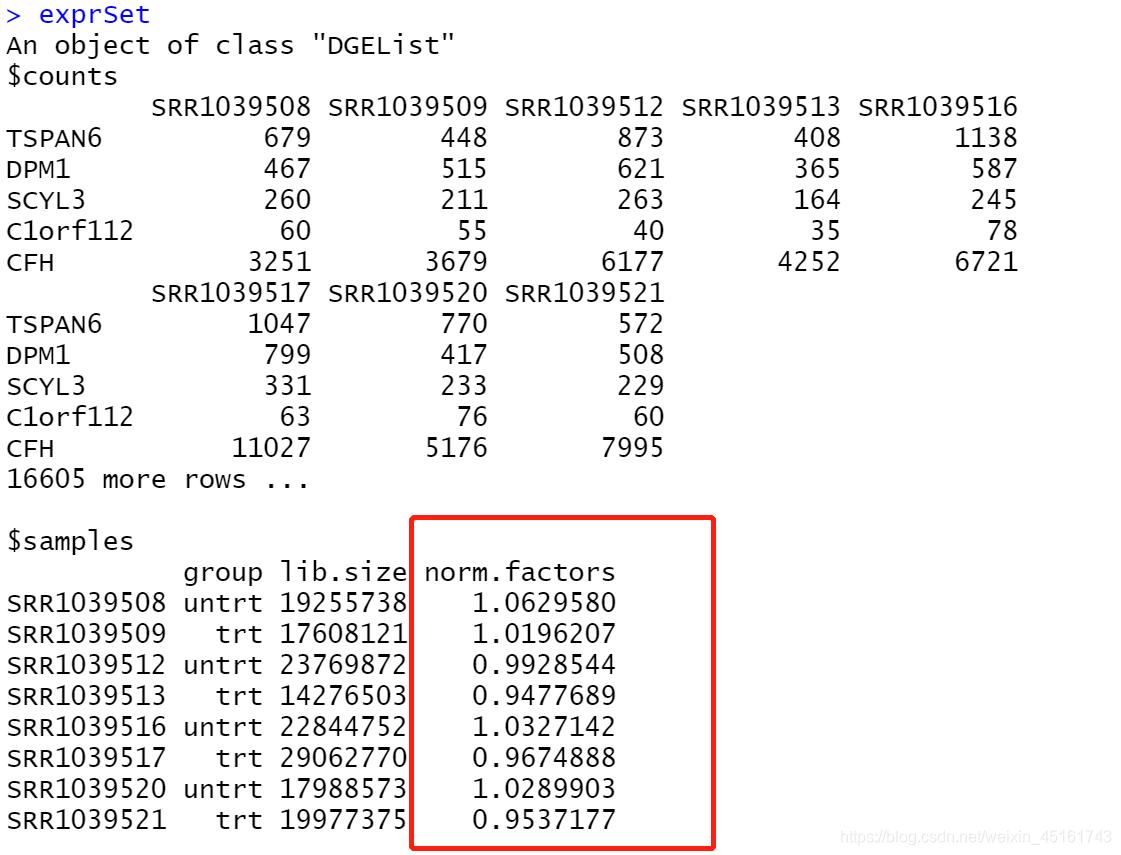
2.TMM标准化
定义: M-值的加权截尾均值。
流程: 选定一个样品为参照,其它样品中基因的表达相对于参照样品中对应基因表达倍数的log2值定义为M-值,随后去除M-值中最高和最低的30%,剩下的M值计算加权平均值,每一个非参照样品的基因表达值都乘以计算出的TMM。
假设: 大部分基因的表达是没有差异的
参考: edgeR的标准化方法
exprSet <- calcNormFactors(exprSet)
3.估算离散值
负二项分布(negative binomial,NB)模型需要均值和离散值两个参数。首先需要根据样本分组,或者说根据实验设计、目的,构建一个分组矩阵。然后使用该分组矩阵,通过加权似然经验贝叶斯(weighted likelihood empirical Bayes)模型,估算每个基因的负二项离散Tagwise(即经验贝叶斯稳健离散值)、Common(即经验贝叶斯稳健离散值的均值)、Trended(即经验贝叶斯稳健离散值的拟合值)
- 构建分组矩阵
design <- (model.matrix(~rev(group_list)))
colnames(design)<-c("Intercept","trt")
ps:
- limma构建的分组矩阵为model.matrix(~0+factor(group_list)),与该分组矩阵的区别
- 差异分析以“分组1”的表达量相较于“分组0”是上调还是下调为准进行统计
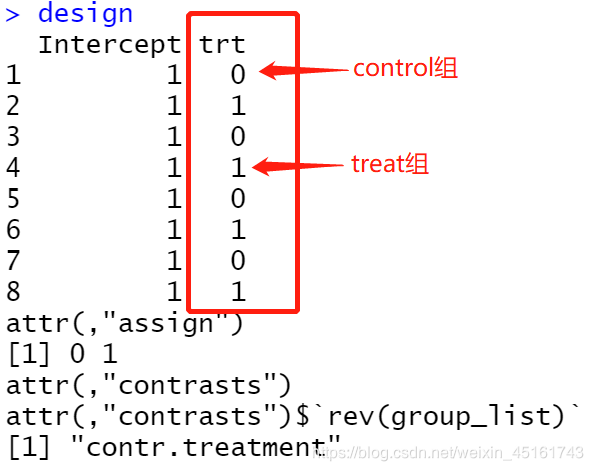
- 这套数据应该为trt是分组1,untrt是分组0,所以需要rev(group_list)来改变factor的顺序
- 估算离散值
exprSet <- estimateDisp(exprSet, design, robust = TRUE)
plotBCV(exprSet)
ps:
- estimateDisp()估算的3个值,其实就是estimateGLMTagwiseDisp()、estimateGLMCommonDisp()和estimateGLMTrendedDisp()的结果组合
- 不指定分组矩阵(单因素实验),则将得到estimateCommonDisp()和estimateTagwiseDisp()的结果组合

4.差异基因分析
- 负二项式广义对数线性模型
如果某个基因的表达值偏离这个分布模型,那么该基因即为差异表达基因
fit <- glmFit(exprSet, design, robust = TRUE)#拟合模型
lrt <- glmLRT(fit) #统计检验
topTags(lrt) #查看p值最显著的差异基因
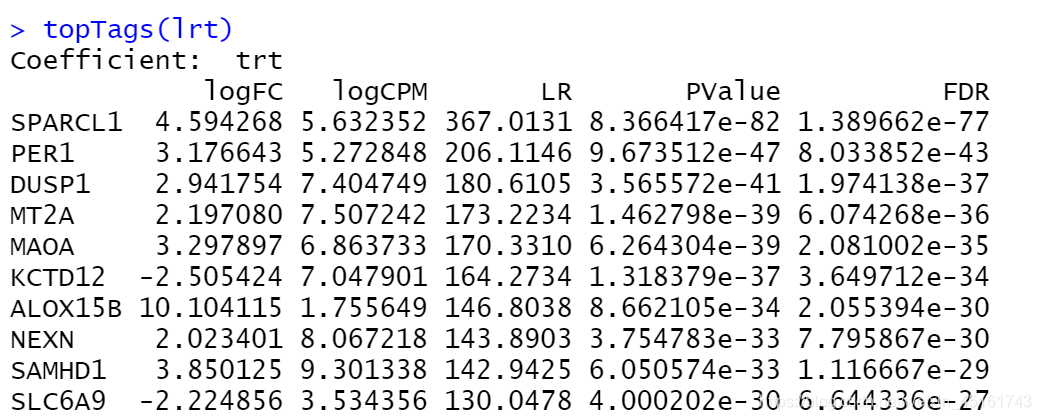
ps:
- LR: 似然比统计
- logFC:根据CPM值计算
- FDR: FDR校正后的p值
#查看默认方法获得的差异基因
dge_de <- decideTestsDGE(lrt, adjust.method = 'fdr', p.value = 0.05)
summary(dge_de)

#提取差异表达基因
allDEG <- topTags(lrt,n = Inf,coef = 'trt-untrt',)
allDEG<-as.data.frame(allDEG)
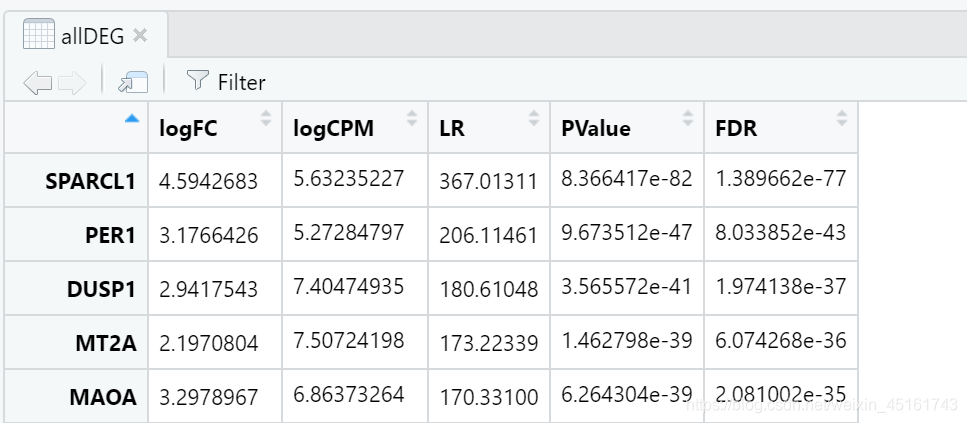
padj = 0.05
foldChange= 1
diff_signif = allDEG[(allDEG$FDR < padj & abs(allDEG$logFC)>foldChange),]
diff_signif = diff_signif[order(diff_signif$logFC),]
save(diff_signif, file = 'edgeR_diff.Rdata')
- 类似然负二项式广义对数线性模型
相较于上一个模型,这个方法更严格一些
fit <- glmQLFit(exprSet, design, robust = TRUE) #拟合模型
lrt <- glmQLFTest(fit) #统计检验
topTags(lrt)
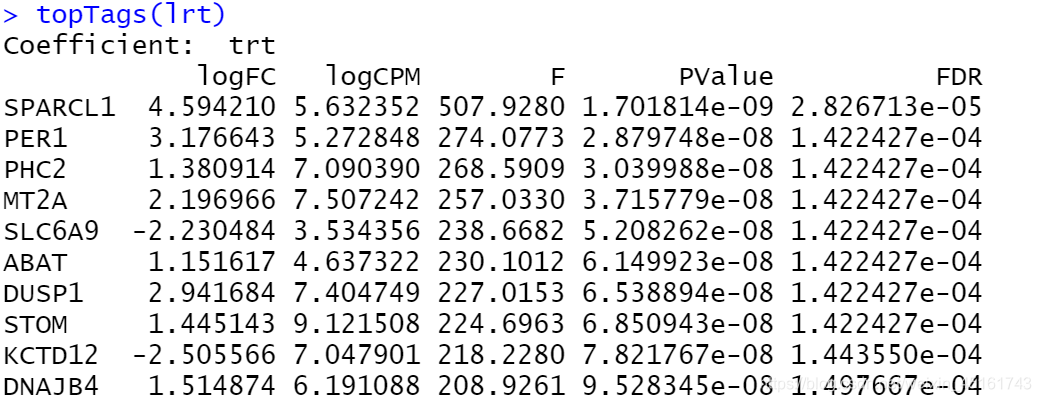
#查看默认方法获得的差异基因
dge_de <- decideTestsDGE(lrt, adjust.method = 'fdr', p.value = 0.05)
summary(dge_de)

- 配对检验
执行两组负二项分布count之间基因均值差异的精确检验,速度较慢
dge_et <- exactTest(exprSet,pair=2:1) #不设置pair时结果相反
topTags(dge_et)
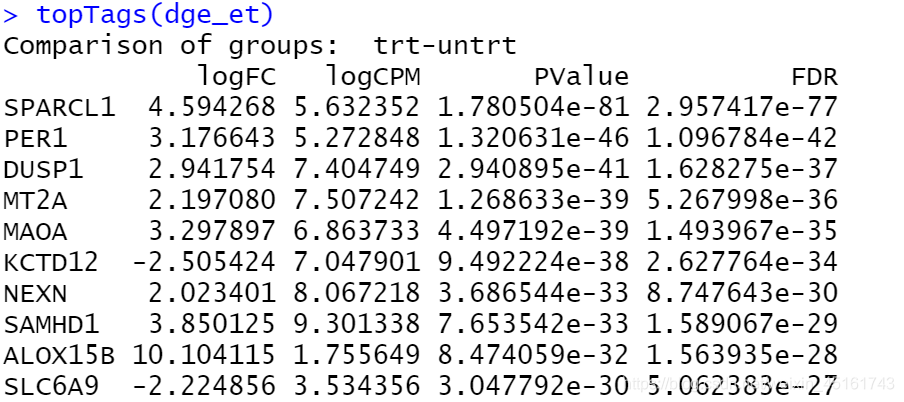
#查看默认方法获得的差异基因
dge_de <- decideTestsDGE(dge_et, adjust.method = 'fdr', p.value = 0.05)
summary(dge_de)
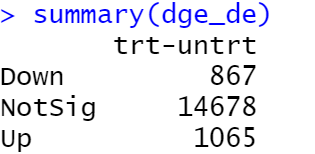
三种方法严格性比较:
类似然负二项式广义对数线性模型>配对检验>负二项式广义对数线性模型


















 2991
2991

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









