







1引言
已经陆陆续续有用户使用一些我写的 R 包了,并且也反馈了一些小问题,这里列出最近的修复结果。
2GseaVis 问题
p 值格式:
有位小伙伴在 github 上提了个不错的建议:
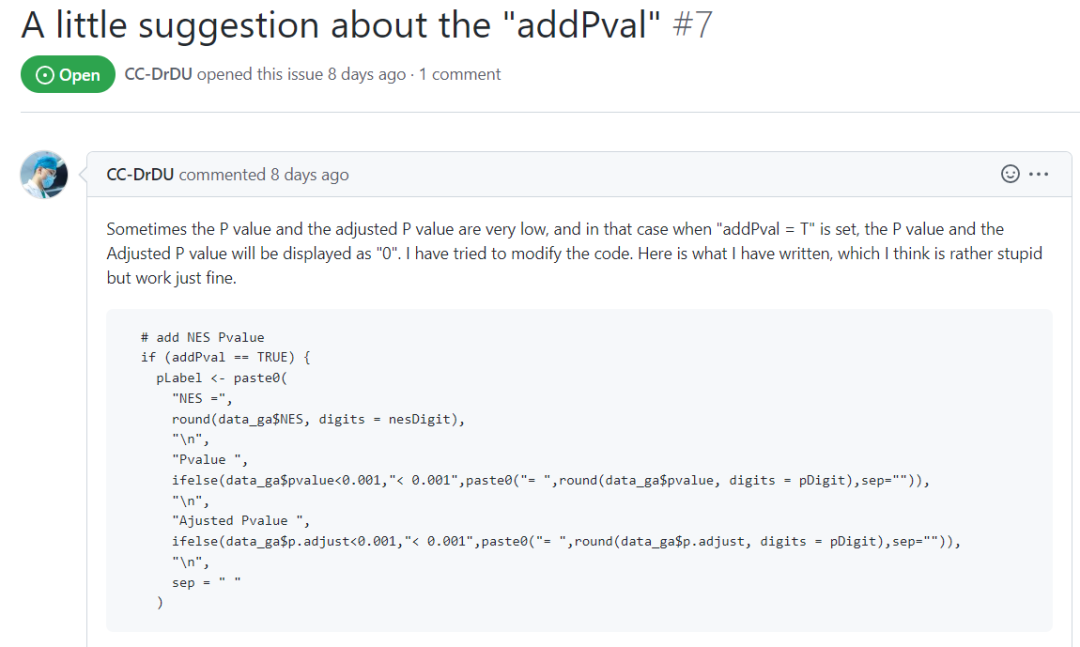
建议对于 p 值太小的时候直接写成小于一个阈值就行了,我之前提供了保留位数参数,这里我就采用了这个建议。
library(GseaVis)
# load data
test_data <- system.file("extdata", "gseaRes.RDS", package = "GseaVis")
gseaRes <- readRDS(test_data)
# df <- data.frame(gseaRes)
# plot
gseaNb(object = gseaRes,
geneSetID = 'GOBP_NUCLEOSIDE_TRIPHOSPHATE_METABOLIC_PROCESS',
subPlot = 2,
addPval = T,
pvalX = 0.85,pvalY = 0.75)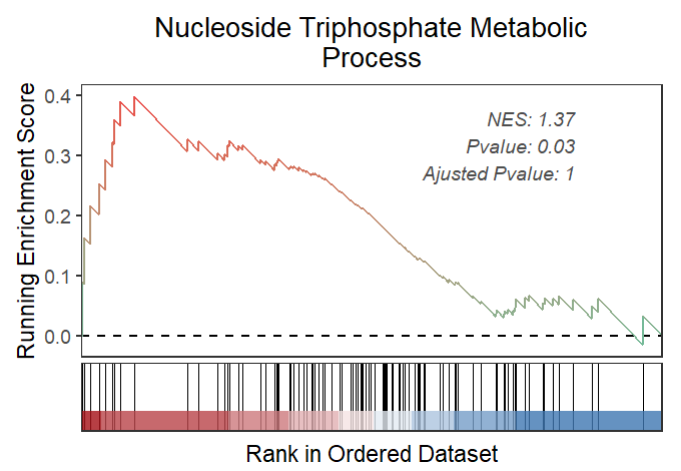
如果遇到非常小的 p 值就写成这样:
gseaNb(object = gseaRes,
geneSetID = 'GOBP_REGULATION_OF_VASCULOGENESIS',
subPlot = 2,
addPval = T,
pvalX = 0.85,pvalY = 0.75)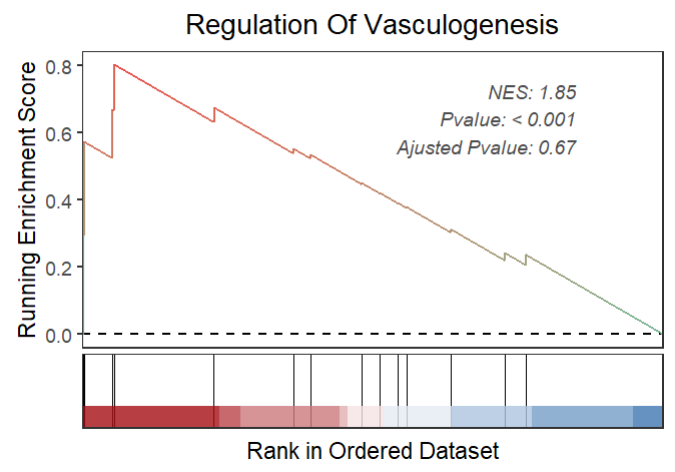
调整曲线粗细
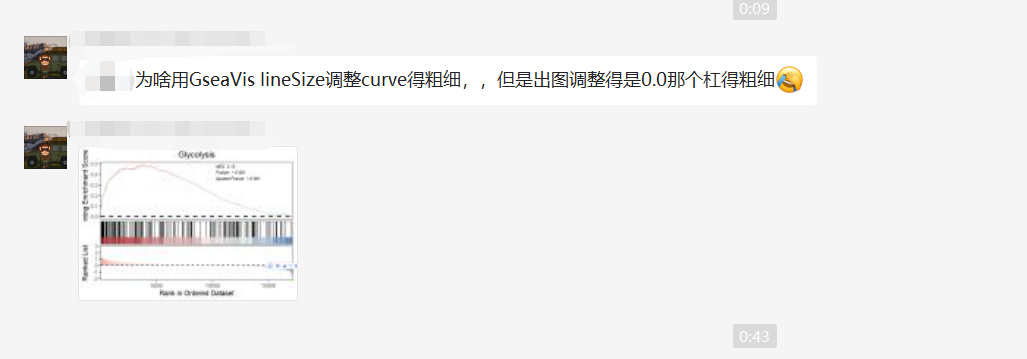
修复了曲线粗细的问题:
gseaNb(object = gseaRes,
geneSetID = 'GOBP_NUCLEOSIDE_TRIPHOSPHATE_METABOLIC_PROCESS',
subPlot = 2,
addPval = T,
pvalX = 0.85,pvalY = 0.75,
lineSize = 2)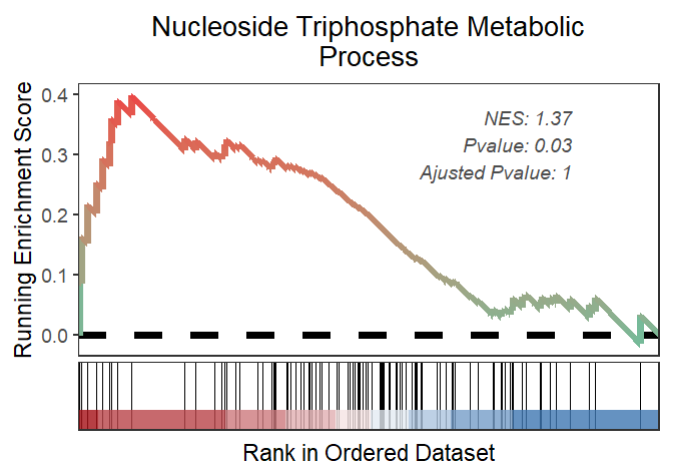
3transplotR 问题
trancriptVis 报错
有小伙伴看了 trancriptVis 可视化 NCBI 的 GTF 文件, 于是拿了自己的 gtf 文件可视化,但是报错了:
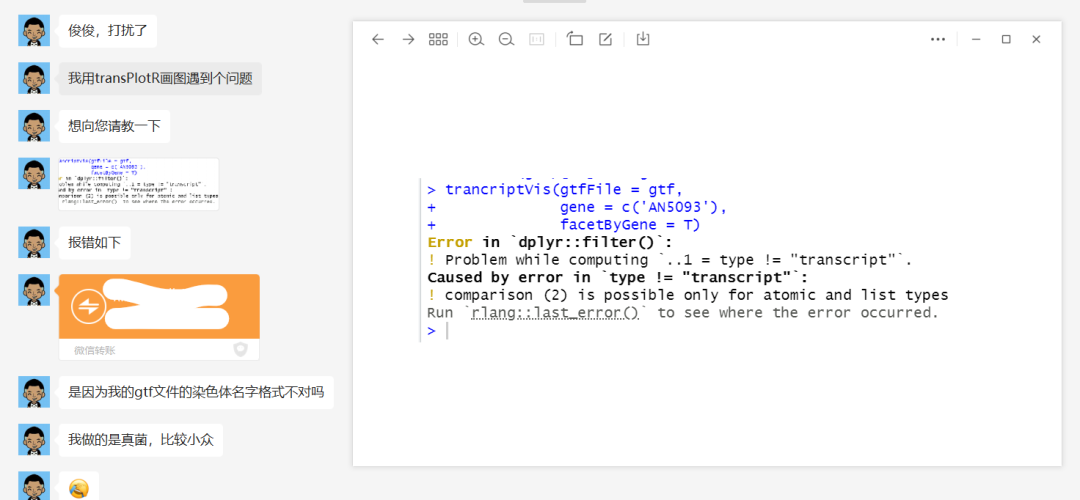
但是做的是 真菌 的物种,可能和人和小鼠还不太一样,仔细检查了一下发现他的 gtf 文件 type 列没有 transcript 的信息,因为真菌大部分是已经基因只有一个转录本,酵母也是这样,所以就没有 transcript 来区分不同的转录本结构了:
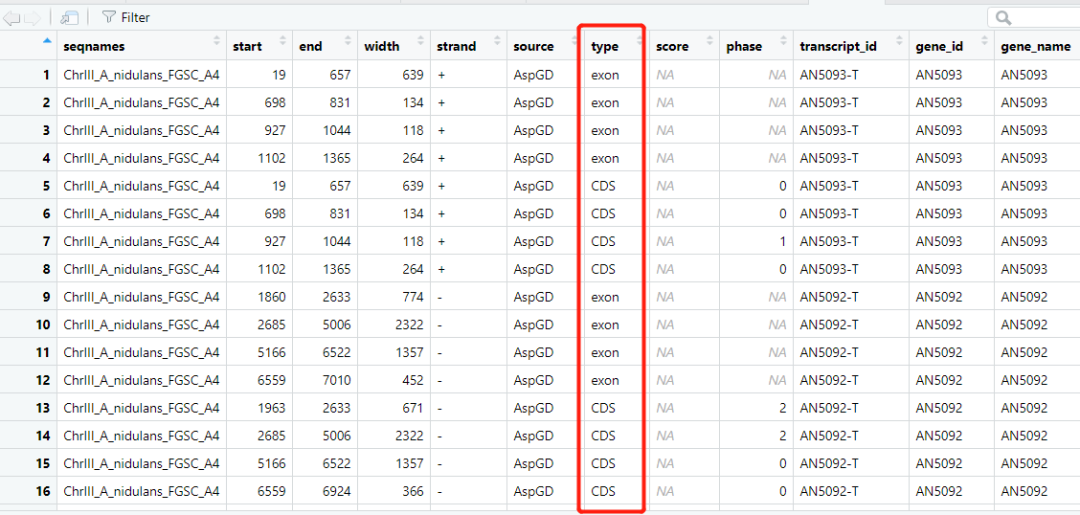
所以我加了一些代码来判断这列是不是有 transcript 关键词,没有的话就加一行信息,起始位置和终止位置则选择该基因的最小起始位置和最大终止位置:

测试:
library(transPlotR)
library(rtracklayer)
gtf <- import('A_nidulans_FGSC_A4_version_s10-m04-r03_features.gtf',format = "gtf") %>%
data.frame()
trancriptVis(gtfFile = gtf,
gene = c('llmE'))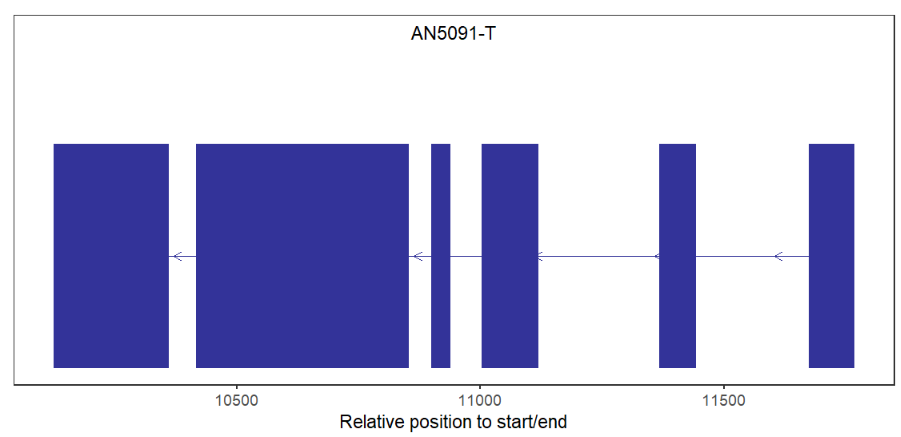
4scRNAtoolVis 问题
箭头坐标位置问题
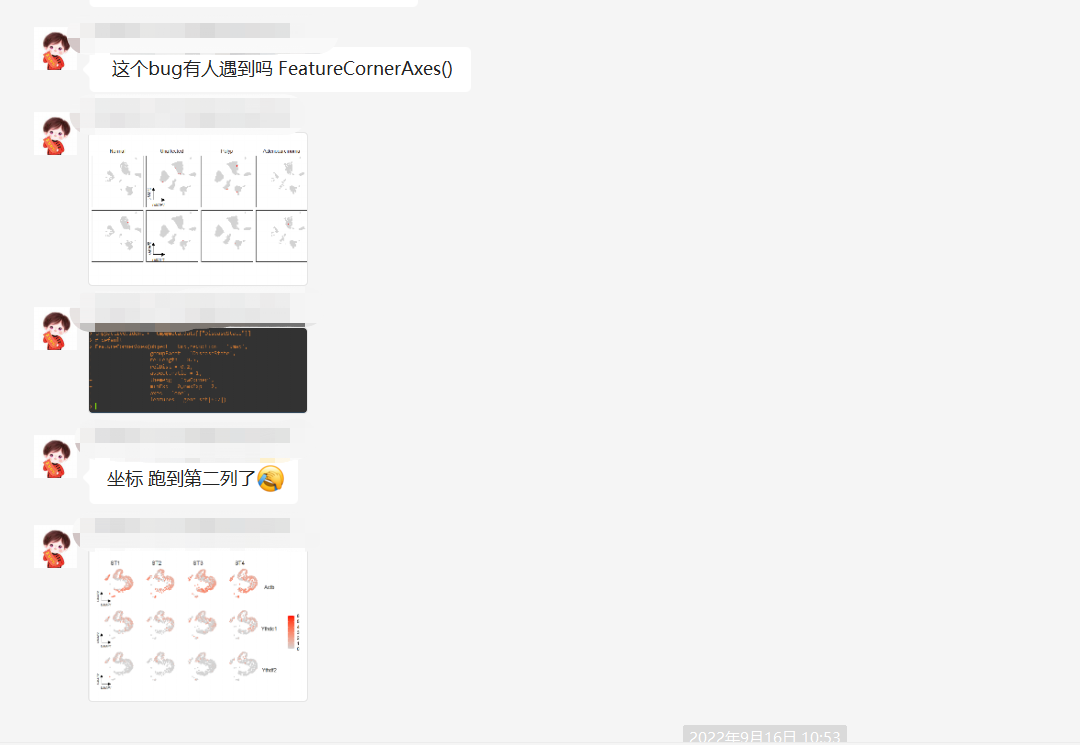
原因是样本里的因子顺序问题,如果没有因子存在,则默认按字符排序取第一个样本的位置添加坐标轴,如果有因子的话没变过来, 所以需要判断用户提供的样本名是否存在因子的情况来做出选择, 修改代码如下:

测试:
library(scRNAtoolVis)
# load test data
test <- system.file("extdata", "seuratTest.RDS", package = "scRNAtoolVis")
tmp <- readRDS(test)
# assign factors
tmp@meta.data$orig.ident <- factor(tmp@meta.data$orig.ident,levels = c('ST3','ST2','ST4','ST1'))
levels(tmp@meta.data$orig.ident)
# [1] "ST3" "ST2" "ST4" "ST1"
# specify corner position
FeatureCornerAxes(object = tmp,reduction = 'umap',
groupFacet = 'orig.ident',
relLength = 0.5,
relDist = 0.2,
aspect.ratio = 1,
features = c("Actb","Ythdc1", "Ythdf2"),
axes = 'one')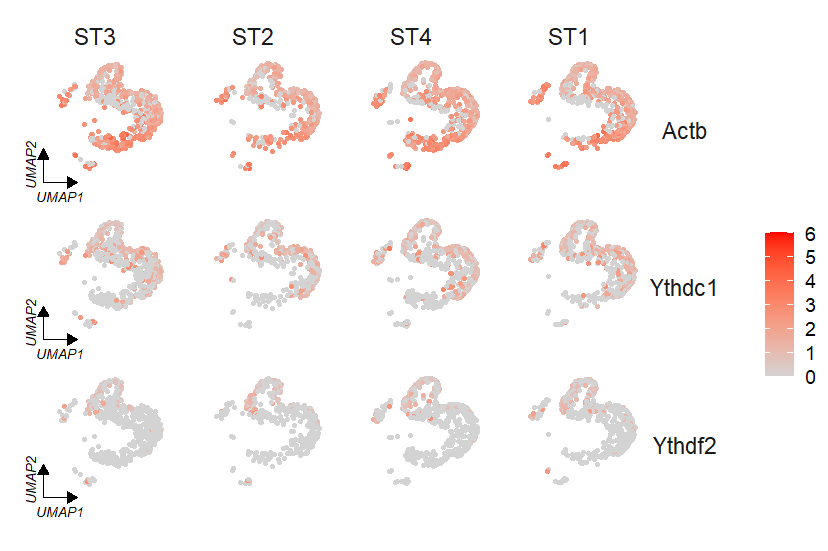
这里我们人为设定顺序为 'ST3','ST2','ST4','ST1', 添加的坐标就自动指定第一个因子的样本名,。
在含有因子的情况下,我们依然可以设定坐标轴位置:
FeatureCornerAxes(object = tmp,reduction = 'umap',
groupFacet = 'orig.ident',
relLength = 0.5,
relDist = 0.2,
aspect.ratio = 1,
features = c("Actb","Ythdc1", "Ythdf2"),
axes = 'one',
cornerVariable = 'ST2')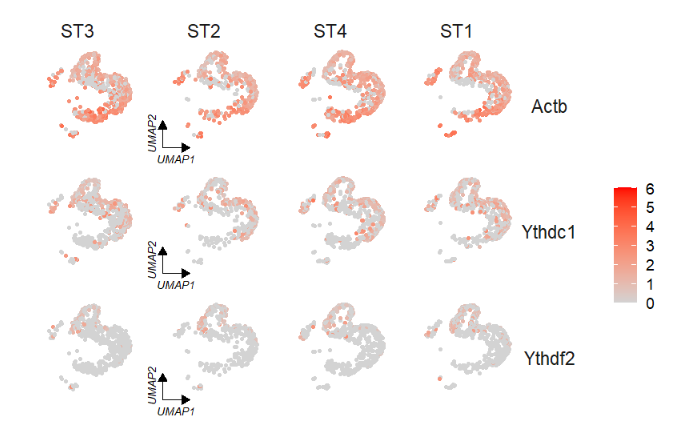
原理则是重新修改了因子的顺序即可。
5结尾
最后感谢大家的支持和肯定,也欢迎留下你的小心心!
https://github.com/junjunlab















 840
840











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









