






1写在前面
最近真是累的不行,今天抽空写一下新的教程,关于人人都会做的GSEA(Gene Set Enrichment Analysis)。
但有时候我们做完GSEA后结果实在太多,无法确定其中重要的生物学意义,难以解释。🤨
本期我们介绍一下GSEAmining包,对我们的GSEA结果做一个瘦身吧,基本原理是:👇
1️⃣ 对参与类似生物过程的基因集应该有
共同的基因。
2️⃣ 对拥有一定数量的共同基因的相似基因集进行功能聚类。
2用到的包
rm(list = ls())
# if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#
# BiocManager::install("GSEAmining")
library(dplyr)
library(GSEAmining)
library(clusterProfiler)
library(msigdbr)
library(org.Hs.eg.db)
3示例数据
这里我们从DOSE包里提取一些基因,作为我们的genelist,假装是我们的输入数据。😙
data(geneList, package="DOSE")
gene <- names(geneList)[abs(geneList) > 2]
# Entrez gene ID
head(gene)

4整理gmt
这里我们用msigdbr包提取一下hallmark,GO和KEGG的基因集。🤒
再也不用去下载gmt文件了,真香!~😂
h_t2g <- msigdbr(species = "Homo sapiens", category = "H") %>%
dplyr::select(gs_name, entrez_gene)
C2_t2g <- msigdbr(species = "Homo sapiens", category = "C2", subcategory = "CP:KEGG") %>%
dplyr::select(gs_name, entrez_gene)
C5_t2g <- msigdbr(species = "Homo sapiens", category = "C5") %>%
dplyr::select(gs_name, entrez_gene)
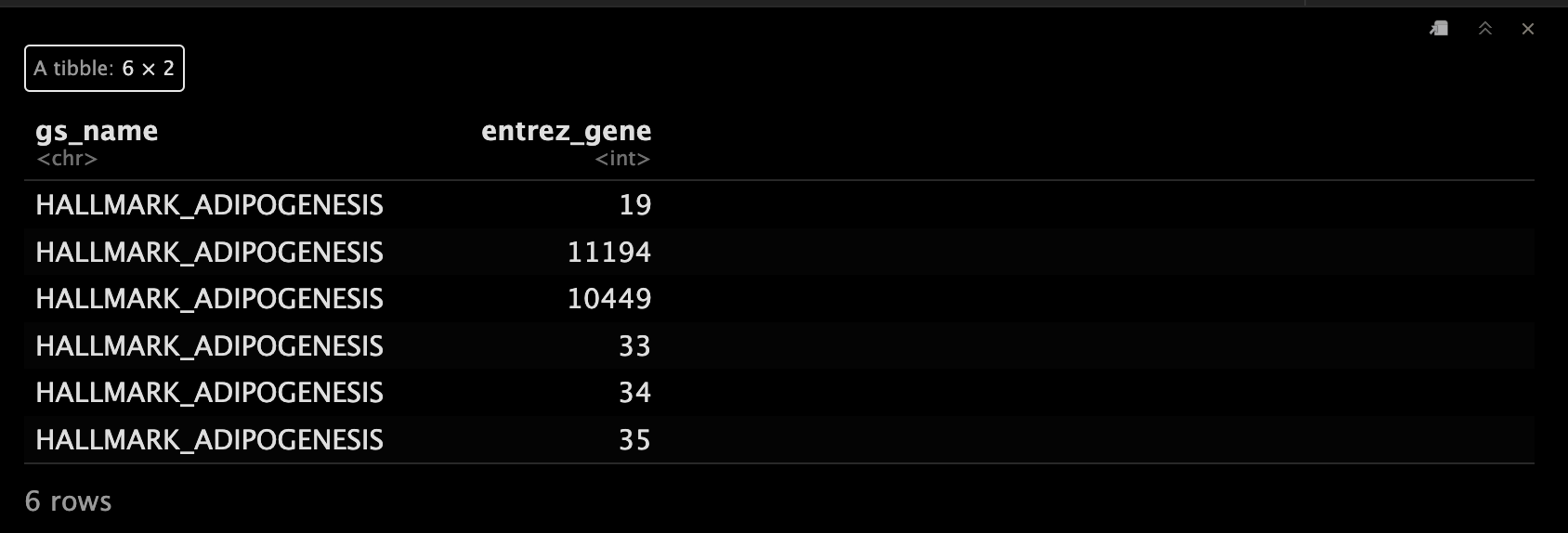
all_t2g <- rbind(h_t2g, C2_t2g, C5_t2g)
head(all_t2g)
5GSEA分析
5.1 开始GSEA
GSEA.res <- GSEA(geneList, TERM2GENE = all_t2g, pvalueCutoff = 0.1, eps = 0)

5.2 将ID转为SYMBOL
GSEA.res <- setReadable(GSEA.res, keyType = "ENTREZID", OrgDb = "org.Hs.eg.db")
dat <- GSEA.res@result
5.3 过滤一下
这里我们设个阈值,过滤一下,实在是太多了。😂
gs.filt <- gm_filter(dat,
p.adj = 0.05,
neg_NES = 2.5,
pos_NES = 2.5)
6聚类
6.1 开始聚类
这里我们进行一下hierarchical clustering,对富集结果进行一下瘦身。🤨
补充一下,这一步是基于core_enrichment的。😷
gs.cl <- gm_clust(gs.filt)
gs.cl

6.2 初步可视化
画个cluster dendrogram吧, 红色 ➡️ positive, 蓝色 ➡️ negative。😙
gm_dendplot(gs.filt,
gs.cl)

6.3 改个颜色
gm_dendplot(gs.filt,
gs.cl,
col_pos = 'orange',
col_neg = 'black',
rect = T,
dend_len = 20,
rect_len = 1)

7分组评估富集结果
这里我们按cluster对各个cluster进行一下深入分析,看看那个term才是最重要的。🤩
7.1 分组分析
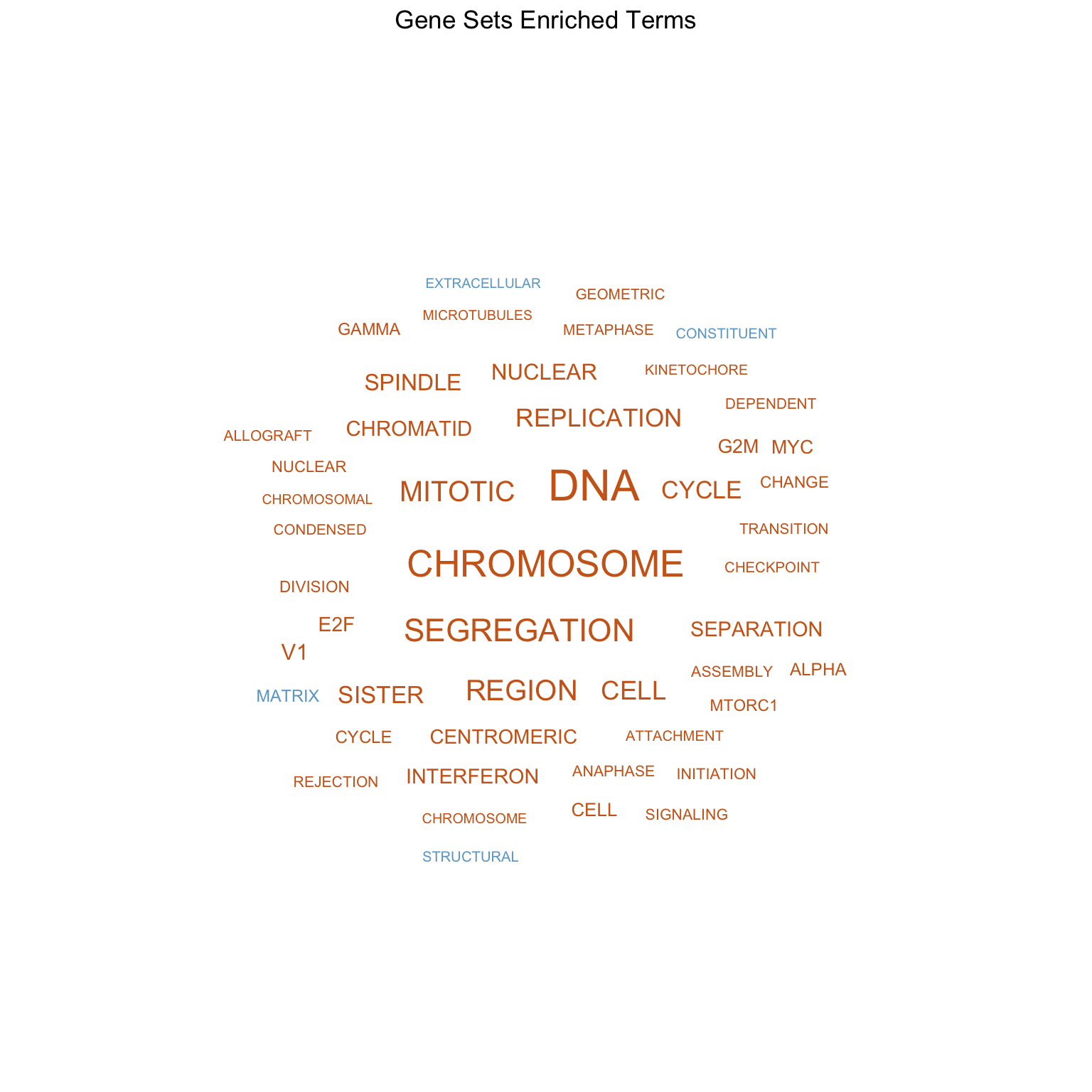
这里我们有4个cluster,看看都是什么term吧。😁
我们用词云的方式展示下结果,越大越有意义。🧐
gm_enrichterms(gs.filt, gs.cl)

7.2 不分组分析
当然你也可以不按cluster分析,全部都放在一起。😂
gm_enrichterms(gs.filt,
gs.cl,
clust = F,
col_pos = 'chocolate3',
col_neg = 'skyblue3')
8分组评估具体基因
对于找到的有意义的基因集,我们也可以看下哪个基因对其贡献最大,在其中起到最重要的作用。😏
gm_enrichcores(gs.filt, gs.cl,
col_pos = 'chocolate3',
col_neg = 'skyblue3')

9如何引用
📍
Arqués O (2022). GSEAmining: Make Biological Sense of Gene Set Enrichment Analysis Outputs. R package version 1.8.0.

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 WGCNA | 值得你深入学习的生信分析方法!~
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布















 2673
2673

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









