






先来感受下仅需1秒的极速出图流程和一键随机修改配色功能,性能遥遥领先:
本次复现的图表来源于Nature正刊上(IF=64.8)题目为《Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma,译:蛋白质组学鉴定早期肝细胞癌新治疗靶点》中的Fig.1。跟着操作,只需要简单的鼠标点点点3步骤,在短暂3分钟内即可轻松复现该图。大家如果有类似数据分析需求可以按照平台上示例数据格式直接复制粘贴替换成自己数据直接分析哦!

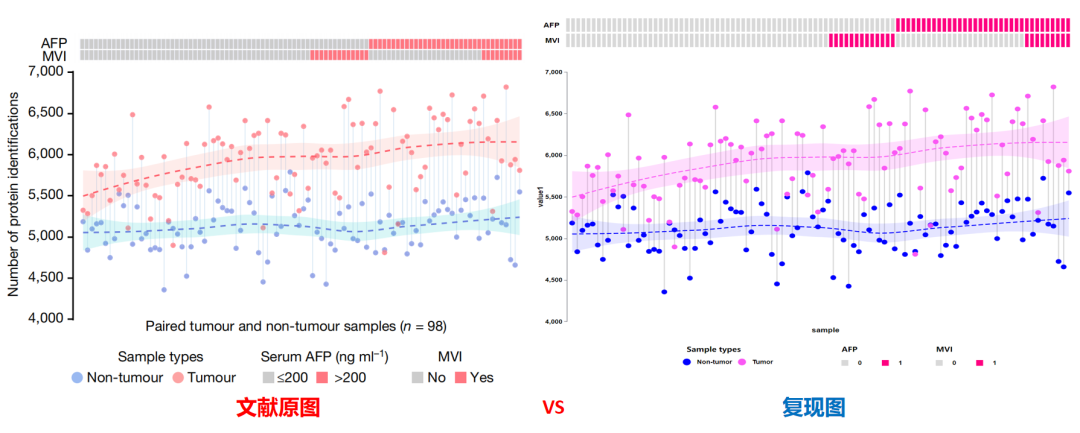
CNSknowall完美一比一复现Nature原图,点的横纵坐标位置和拟合曲线轨迹走向均与原文丝毫不差,另外采用经典的红蓝CP对配色进行了加深优化以凸显对比!
复现目标图片介绍
--- ·配对哑铃组合图 · ---
配对哑铃组合图由两部分构成:
第一部分为分类变量的格子热图——分类格子热图是一种用于可视化两个分类变量之间关系的图表,用颜色表示类别,并附带标签;
第二部分为带有拟合曲线的配对哑铃图,包含的元素有:
(1)配对哑铃图:哑铃图(Dumbbell Plot)又称DNA图,顾名思义,这类图形看起来形似哑铃和DNA,其特点是是在两个时间点或条件之间用线段连接两个点,形成一个类似哑铃的形状,从而清晰地显示出变化的趋势。配对哑铃图是一种用于可视化两个指标/变量、两个组别/群体/类别、两个不同时间点、两个条件或两种状态之间的变化差异或趋势的图表类型,适用于两组以上数据的比较,有助于突出差异或变化趋势。
(2)拟合曲线:为了更好地展示变量之间的趋势,数据使用loess方法进行拟合得到拟合曲线,拟合曲线的阴影表示95%的置信区间。
(3)点:每个配对有上下两个点代表具体的观测值。这有助于更详细地了解数据的分布。
文章从110例早期肝细胞癌(HCC)患者中选择手术切除的原发性肿瘤组织和配对的非肿瘤肝组织作为实验样本,研究通过定量蛋白质组学技术对98对肿瘤和非肿瘤样本进行蛋白质鉴定,并统计了样本的α-胎蛋白的水平(AFP)和显微血管浸润(MVI)是否为阳性,最终得到了待作图的数据。该数据的可视化结果如下:
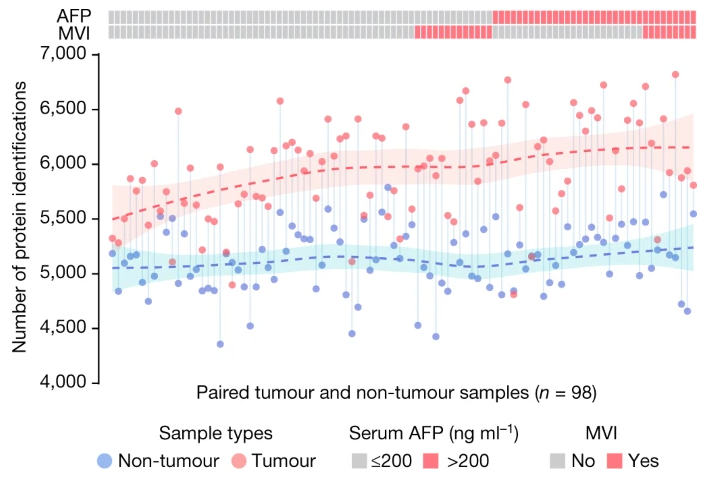
原文Fig.1
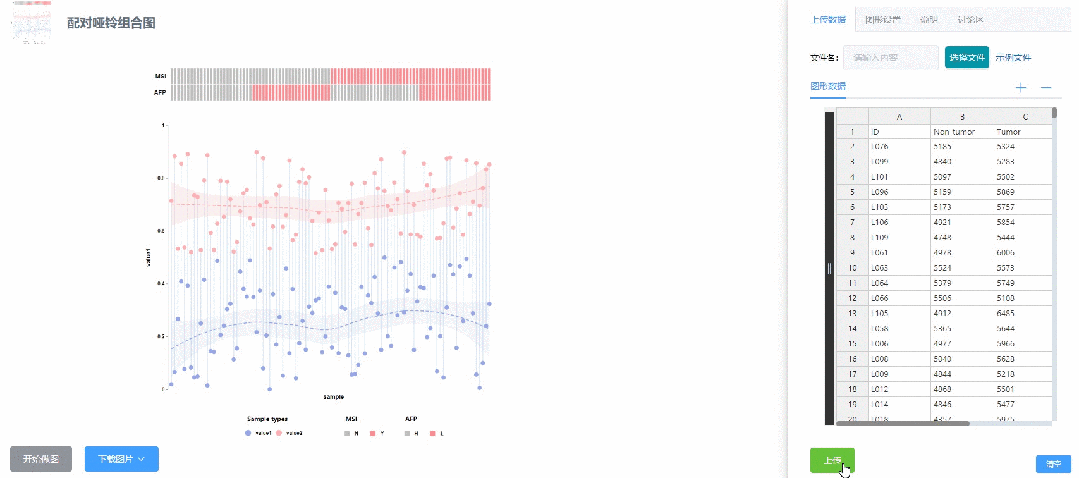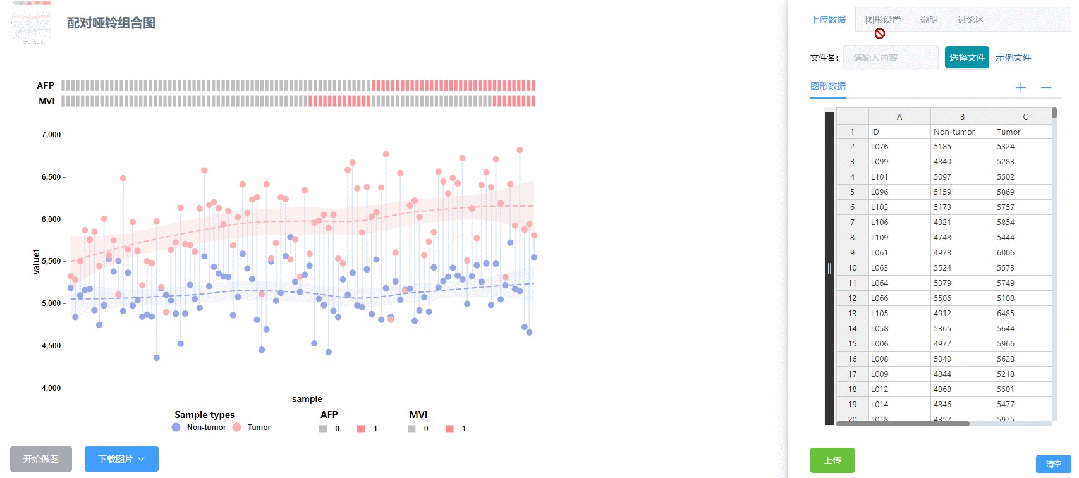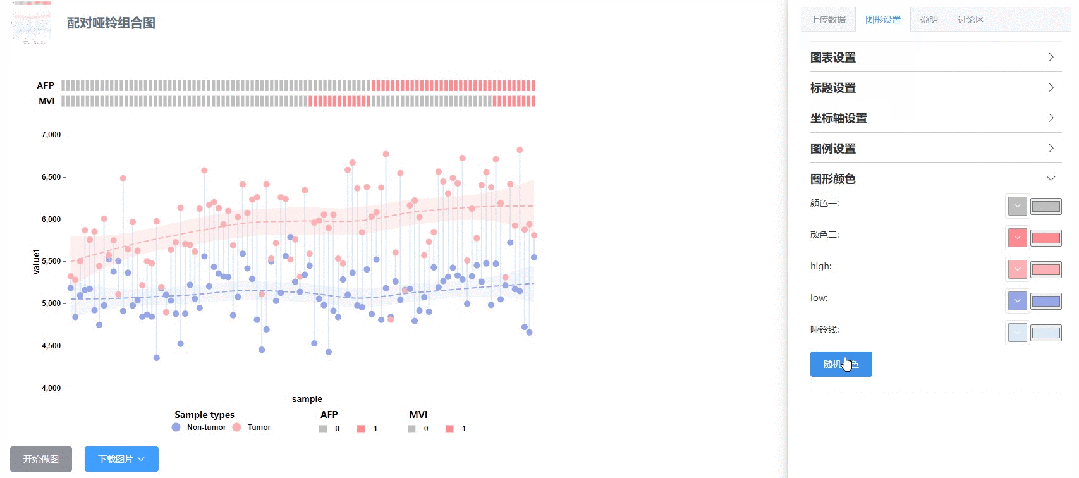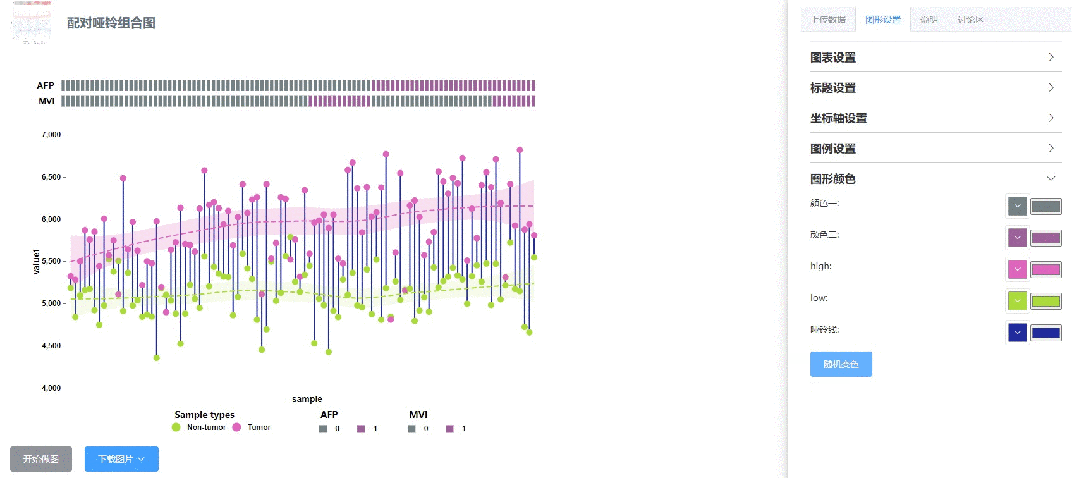
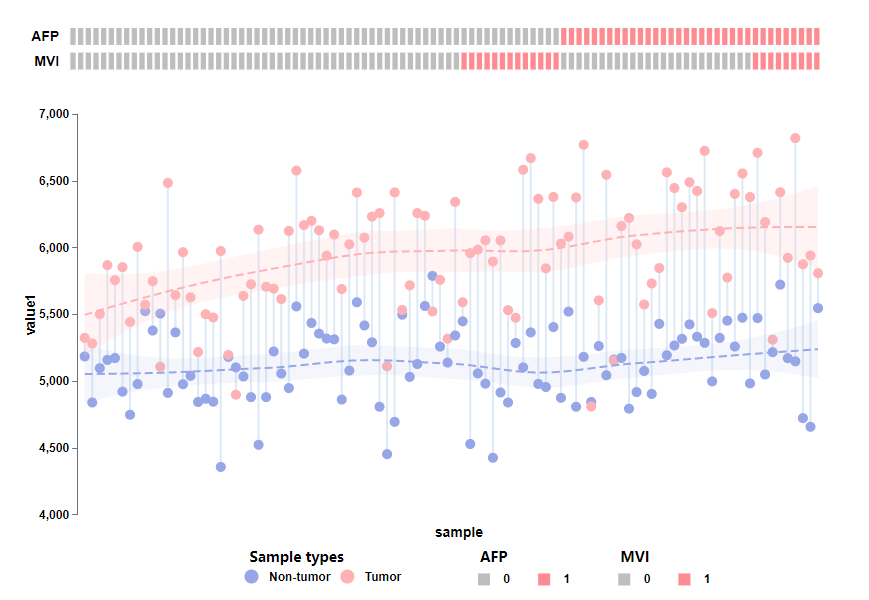
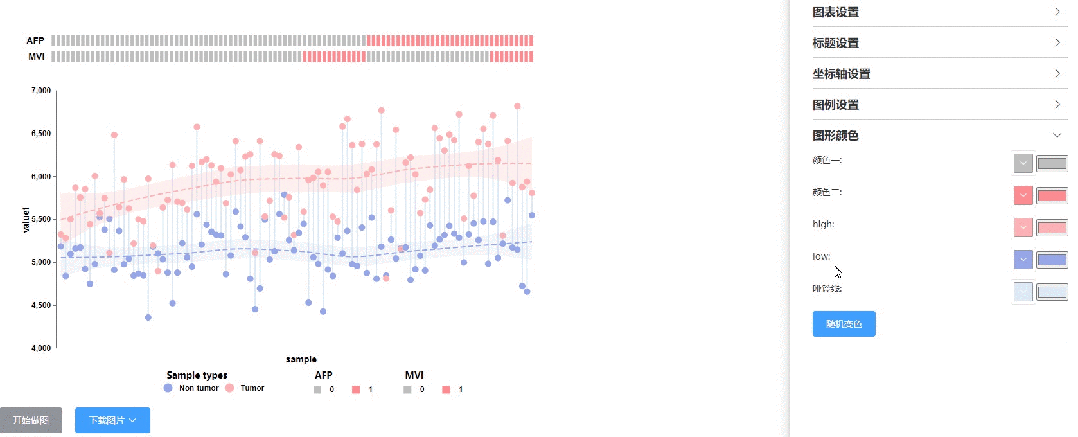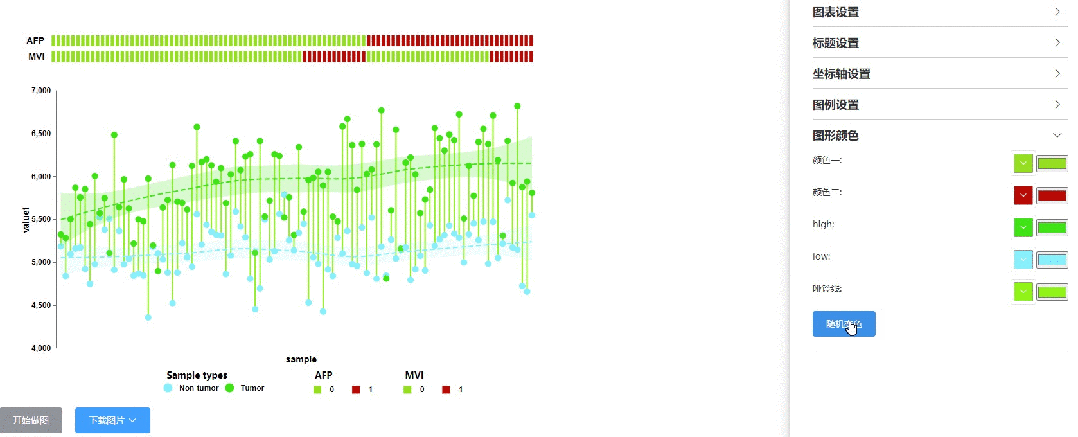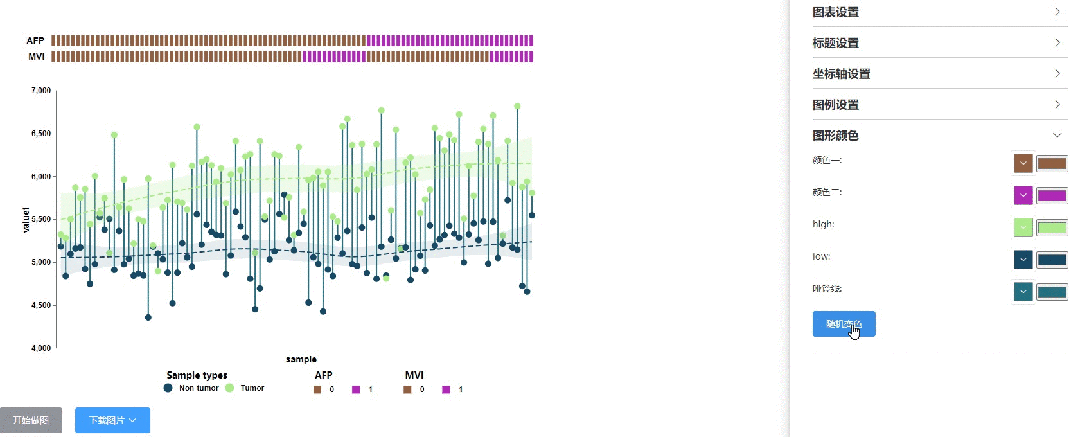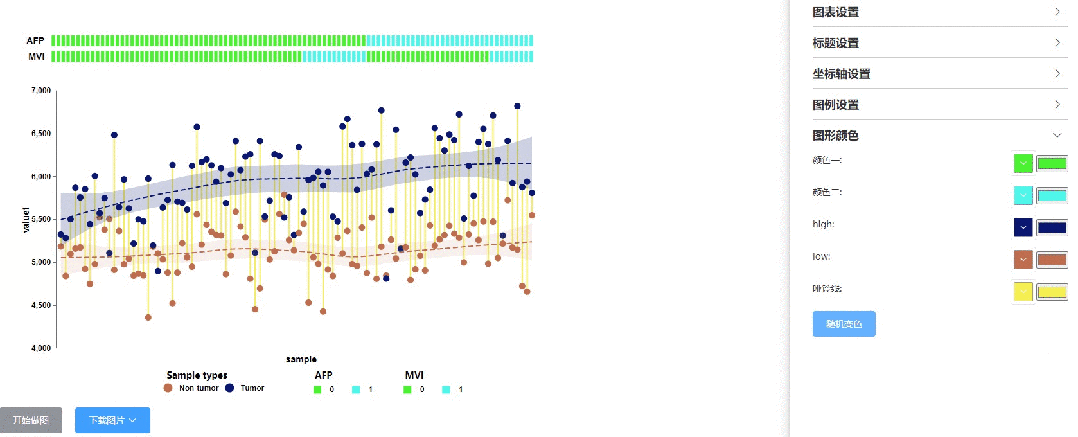
该结果的哑铃图部分展示了每对配对肿瘤与非肿瘤样本中检测出的蛋白质样本数量对比——肿瘤样本明显高于非肿瘤样本;并使用lasso回归分别拟合了肿瘤(红色,n = 98)和非肿瘤(蓝色,n = 98)样本中蛋白质数量的分布,让两组蛋白质数据的特征和趋势更加明显,强烈凸显出肿瘤样本蛋白数量更高;而在分类格子热图中,AFP(α-胎蛋白水平)大于200的样本和MVI+(显微血管浸润阳性)的样本表示为红色,它们明显少于AFP水平低和MVI-的样本。总的来说,该图是对早期肝细胞癌病例中定量蛋白质鉴定的概述,证明了早期肝癌在蛋白质数量水平方面的异质性。当然,该图也可以应用于任何多样本数量统计、分组分类、数据特征展示等分析结果可视化中,具有广泛的应用场景。话不多说,直接看如果3分钟内3步骤0代码画出100%一比一复现该图:
详细步骤
配对哑铃组合图复现
1
登录CNSknowall平台
点击工具链接:
http://cnsknowall.com/#/HomePage
直接微信扫码登录即可免费使用,进入“数据分析模块”——“高级分析”——“差异分析”,找到配对哑铃组合图,点击进入配对哑铃组合图分析界面。

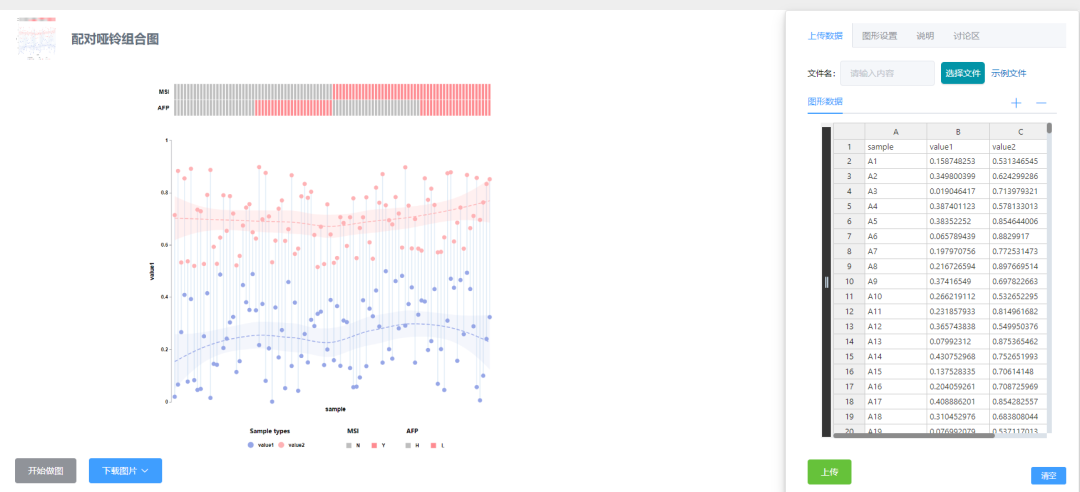
CNSknowall配对哑铃组合图可视化界面
2
数据下载
从原文下载Source Data数据,Nature原文链接:
文献地址:https://www.nature.com/articles/s41586-019-0987-8
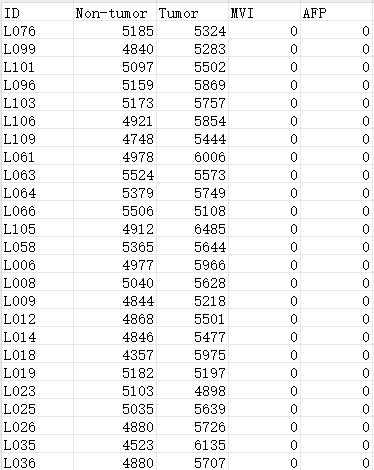
下载数据Souce Data Fig.1,原文提供的Souce Data Fig.1如下:
第一列为样本名(患者),第二列为从非肿瘤样本中鉴定出的蛋白质数量,第三列为从配对肿瘤样本中鉴定出的蛋白质数量,第四列表示样本中是否呈显微血管浸润阳性,若‘是’则表示为‘1’,第五列表示样本的α-胎蛋白水平是否大于200,若‘是’则表示为‘1’。
3
数据上传
将表格数据清空后,将上述Souce Data Fig.1中的数据直接复制粘贴到平台右侧表格中,点击左下方上传选项
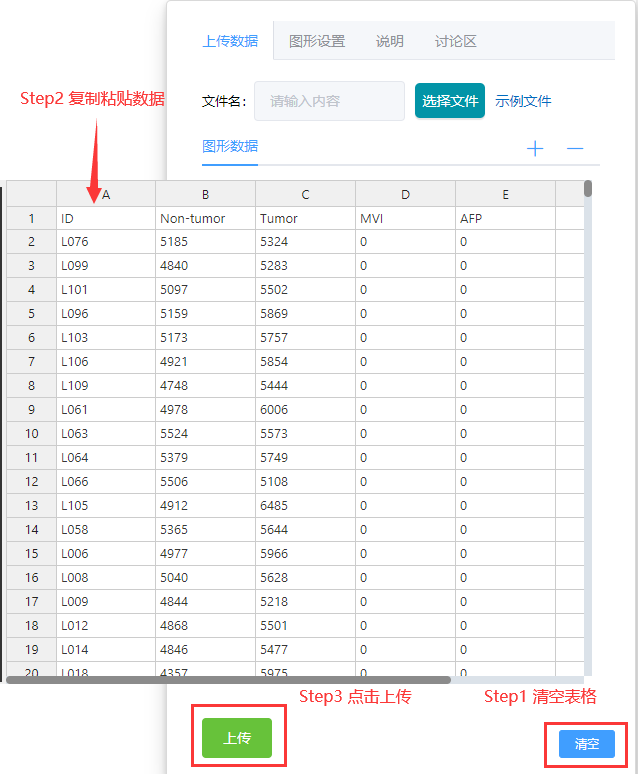
上传成功后,页面上方会显示弹窗信息,“已上传成功”。

最后点击点击页面左下角“开始做图”,等待平台运行。

<1秒即可得到的运行结果,如下:
4
图形细节设置
01
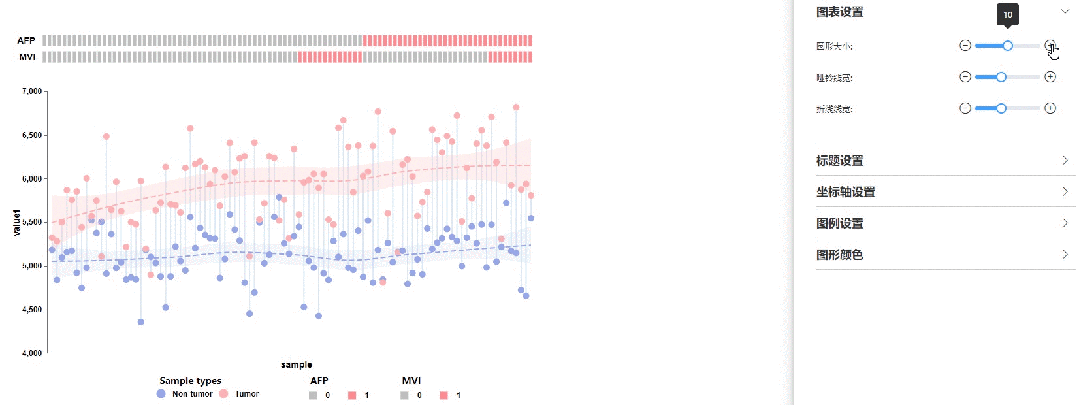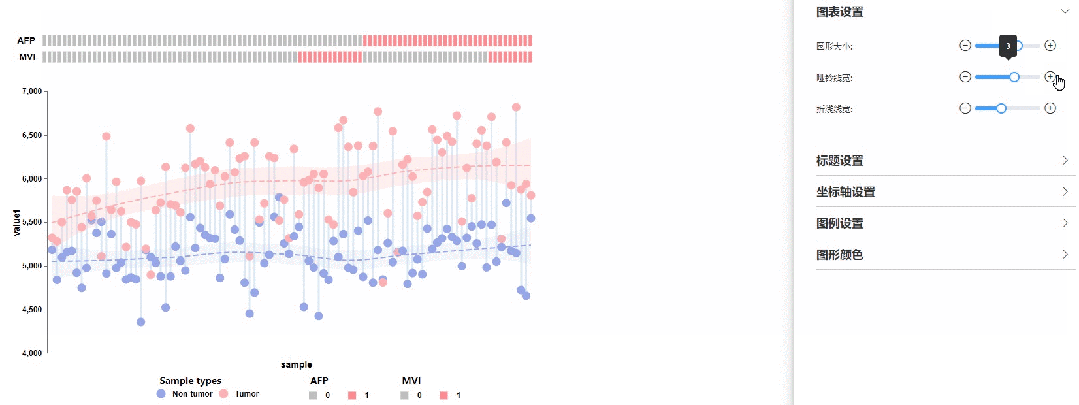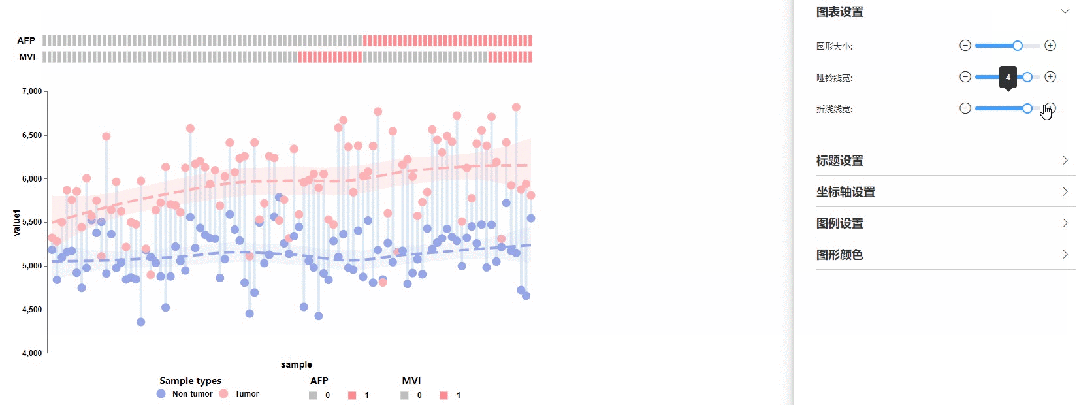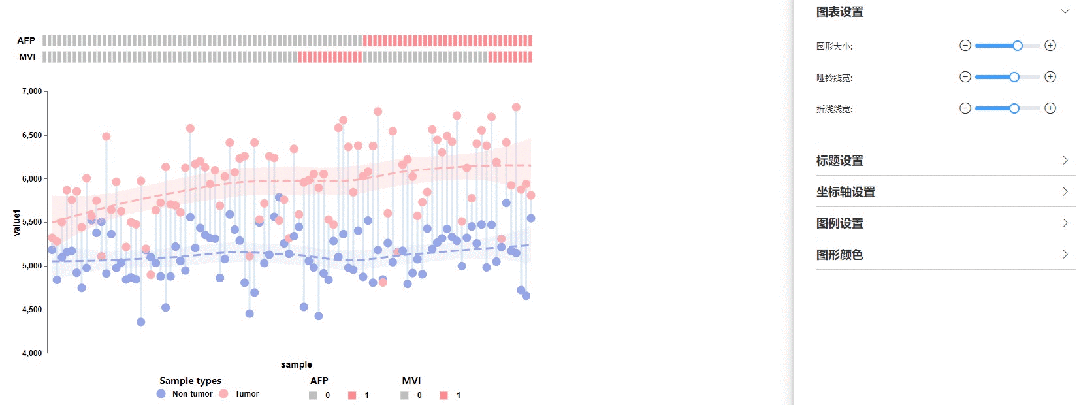
图表设置
修改圆点大小、哑铃线宽以及拟合曲线线宽
02
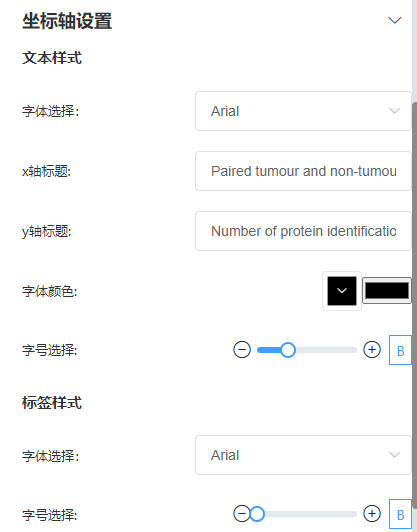
坐标轴设置
x轴标题改为‘Paired tumour and non-tumour samples (n = 98)’,y轴标题改为‘Number of protein identification’,并略微调大字号。
03
图例设置
5
图形颜色设置
方式1:全网首创的一键随机变色模式,遥遥领先:
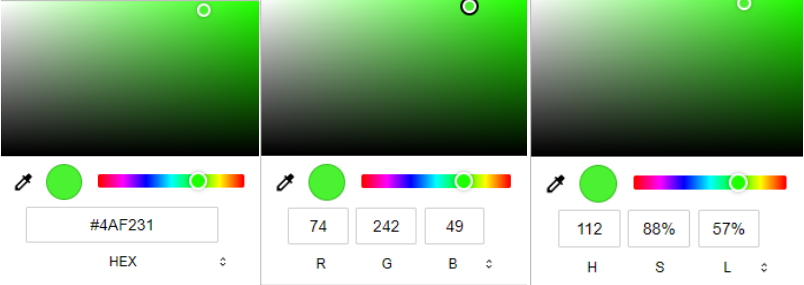
方式2:输入精准的颜色参数
方式3:首次将取色器用于医学数据分析,一键复制相中文献上的配色风格:
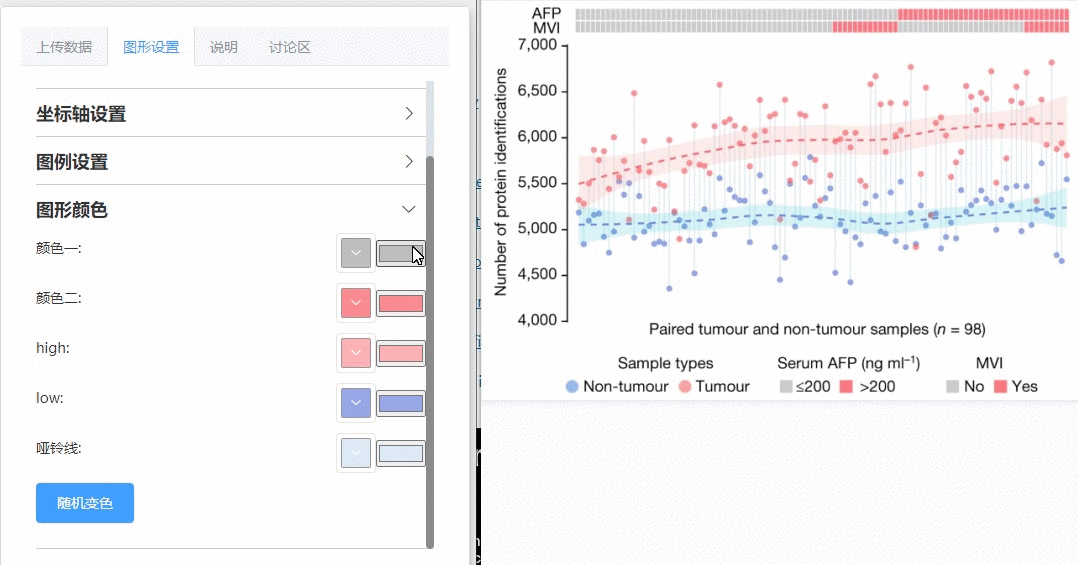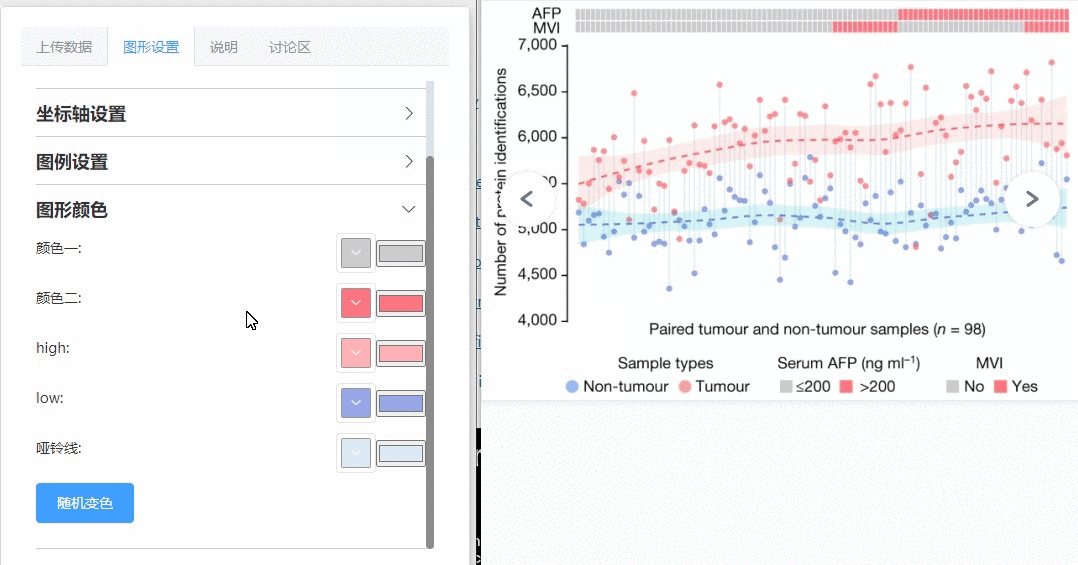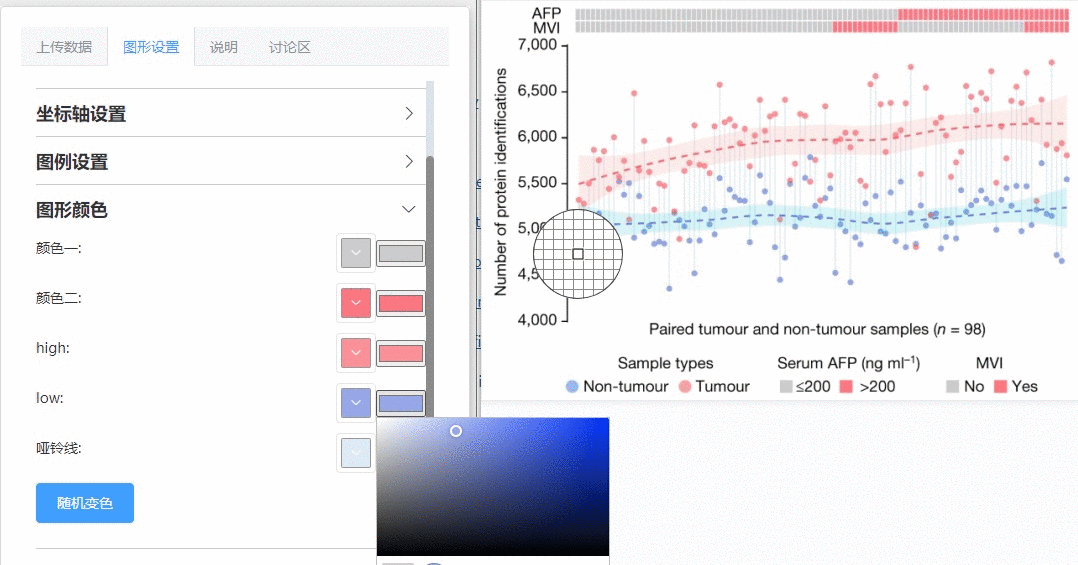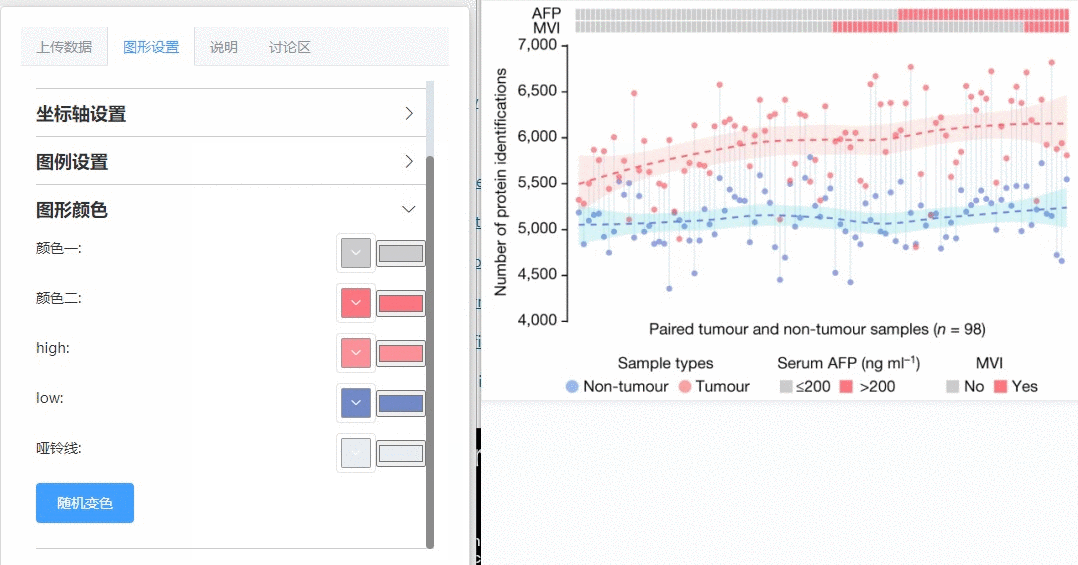
6
输出结果
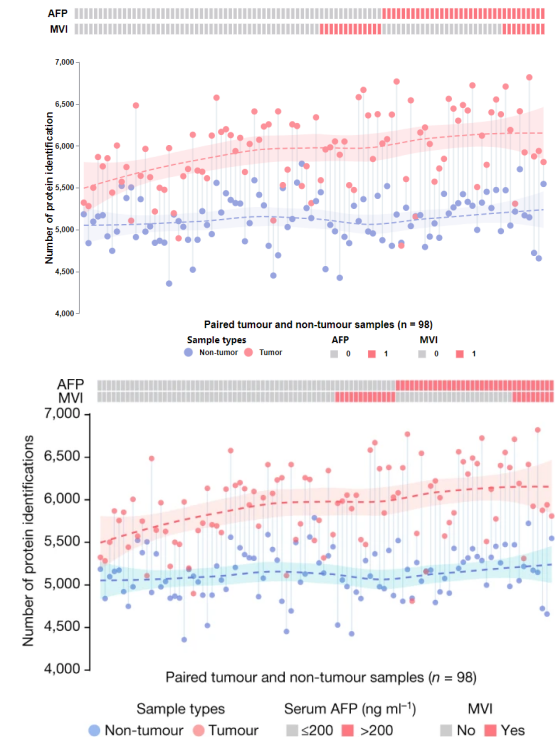
再次看下复现结果图与文献原图对比(上图是CNSknowall复现结果,下图是原文结果),每个点的横纵坐标、拟合曲线形状和走势均一比一100%完美复现:
最后直接下载出可满足SCI发表像素(>300dpi)的图片。本工具有四种图片格式可供下载,可根据需要,自行选择。以下载pdf格式的图片为例,点击“下载图片”--“Download PDF",即可完成下载,下载后的矢量pdf可以进一步编辑各个细节如字体大小和格式、移动标签位置等。
END

点击关注我们,用最短的时间和最高的效率学习更多数据分析方法!
加入我们的官方群咨询平台使用方法,高效学习更多数据分析方法,会晤道友!

免费注册登录CNSknowall
--一次性收藏120个皆可一键出图的高级通用生信工具--
同时收藏42个柱状图+23个饼图+其余70个各类常用图表
写在后面:AI时代已来,您需要非同以往的更强数据分析工具
CNSknowall (中文:CNS万事通)平台是今年1月份新上线的一款专门针对医学领域的创新型免费在线数据分析云平台,和目前常用的数据分析工具如SPSS、Origin、GraphPad Prsim和R语言相比,CNSknowall在数据上传、配色修改和参数调整等各方面做出了一系列重大创新,各项性能遥遥领先,几乎没有任何学习成本(包括时间成本和金钱成本),甚至优于GPT(毕竟GPT不是专门的数据分析工具)。您只需要简单的套用平台提供的固定数据格式复制粘贴替换成自己的数据,鼠标点点点就可以完成CNS级别的高水平图表制作,可以让不擅长或没接触过数据分析的人以最短的时间内快速建立医学数据分析的基本思维,以最快的速度掌握各种数据分析技能,帮助大家在数据分析上节省大量宝贵的时间,从而可以把时间和精力用在更重要的事情比如查阅文献和设计研究思路方案等,提高文章发表速度,减缓毕业焦虑,赋能职业生涯,开启科研天骄之路!
CNSknowall 首页
很多时候知道自己要画什么图往往比会画什么图更重要
平台包含300个数据分析模块,您可以快速找到能让自己数据价值最大化的分析方法

















 152
152

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









