




Free Energy Calculations: Methane in Water
自由能计算:甲烷在水中的情况
Justin A. Lemkul, Ph.D. 贾斯汀·A·勒姆库尔,博士
Virginia Tech Department of Biochemistry
弗吉尼亚理工大学生物化学系
| This tutorial will guide the user through the process of calculating a simple free energy change, the decoupling removal of van der Waals interactions between neutral methane and a box of water. This quantity was previously calculated by Shirts et al. in a systematic evaluation of free energies of solvation of amino acid side chains using different atomistic force fields. This tutorial requires a GROMACS version in the 2018.x series. |
Step One: Theory 第一步:理论
| This tutorial will assume you have a reasonable understanding of what free energy calculations are, the different types that exist, and the underlying theory of the technique. It is neither practical nor possible to provide a complete education here. Instead, I will focus this tutorial on practical aspects of running free energy calculations in GROMACS, highlighting relevant theoretical points as necessary throughout the tutorial. I will provide here a list of useful papers for those who are new to free energy calculations. Do not consider this list exhaustive. It should also not supplant proper study of statistical mechanics and the many books and papers that have been written on related topics.
The objective of this tutorial is to reproduce the results of a very simple system for which an accurate free energy estimate exists. The system of choice (methane in water) is one dealt with by Shirts et al. in a systematic study of force fields and the free energies of hydration of amino acid side chain analogs. The complete publication can be found here. This tutorial will assume you have read and understood the broader points of this paper. Rather than use the thermodynamic integration approach for evaluating free energy differences, the data analysis conducted here will utilize the GROMACS "bar" module, which was introduced in GROMACS version 4.5 (and previously called g_bar). It uses the Bennett Acceptance Ratio (BAR, hence the name of the module) method for calculating free energy differences. The corresponding paper for BAR can be found here. Knowledge of this method is also assumed and will not be discussed in great detail here. Free energy calculations have a number of practical applications, of which some of the more common ones include free energies of solvation/hydration and free energy of binding for a small molecule to some larger receptor biomolecule (usually a protein). Both of these procedures involve the need to either add (introduce/couple) or remove (decouple/annihilate) the small molecule of interest from the system and calculate the resulting free energy change. There are two types of nonbonded interactions that can be transformed during free energy calculations, Coulombic and van der Waals interactions. Bonded interactions can also be manipulated, but for simplicity, will not be addressed here. For this tutorial, we will calculate the free energy of a very simple process: turning off the Lennard-Jones interactions between the atomic sites of a molecule of interest (in this case, methane) in water. This quantity was calculated very precisely by Shirts et al. and thus it is the quantity we seek to reproduce here. |
Step Two: Examine the Topology
第二步:检查拓扑结构
| Download the coordinate file and topology for this system. These files were provided as part of David Mobley's tutorial for this system (which is no longer online), and are the original files (modified slightly for compatibility with recent GROMACS versions) used by Michael Shirts in the paper referenced on the previous page. The system contains a single molecule of methane (called "ALAB" in the coordinate file, for the β-carbon of alanine) in a box of 596 TIP3P water molecules. Looking into the topology, we find: ; Topology for methane in TIP3P
#include "oplsaa.ff/forcefield.itp"
[ moleculetype ]
; Name nrexcl
Methane 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
1 opls_138 1 ALAB CB 1 0.000 12.011
2 opls_140 1 ALAB HB1 2 0.000 1.008
3 opls_140 1 ALAB HB2 3 0.000 1.008
4 opls_140 1 ALAB HB3 4 0.000 1.008
5 opls_140 1 ALAB HB4 5 0.000 1.008
[ bonds ]
; ai aj funct c0 c1 c2 c3
1 2 1
1 3 1
1 4 1
1 5 1
[ angles ]
; ai aj ak funct c0 c1 c2 c3
2 1 3 1
2 1 4 1
2 1 5 1
3 1 4 1
3 1 5 1
4 1 5 1
; water topology
#include "oplsaa.ff/tip3p.itp"
[ system ]
; Name
Methane in water
[ molecules ]
; Compound #mols
Methane 1
SOL 596
You will note that all charges are set to zero. There is a practial reason behind this setup. Normally, charge interactions between the solute and water are turned off prior to the van der Waals terms. If charge interactions are left on when Lennard-Jones terms are turned off, positive and negative charges would be allowed to approach one another at infinitely close distances, resulting in a very unstable system that would probably just blow up. The procedure in this tutorial essentially assumes that charges have been properly been turned off prior to this point, and we will be turning off only van der Waals interactions between the solute and solvent. Note that in previous versions of GROMACS, the contents of the |
Step Three: The Workflow 第三步:工作流程
| The free energy change of transforming a system from state A to state B, ΔGAB, is a function of a coupling parameter, λ, which indicates the level of change that has taken place between states A and B, that is, the extent to which the Hamiltonian has been perturbed and the system has been transformed. Simulations conducted at different values of λ allow us to plot a ∂H/∂λ curve, from which ΔGAB is derived. The first step in planning free energy calculations is how many λ points will be used to describe the transformation from state A (λ = 0) to state B (λ = 1), with the goal of collecting adequate data to exhaustively sample phase space and produce a reliable ∂H/∂λ curve.
For decoupling Coulombic interactions, which depend linearly upon λ, an equidistant λ spacing from 0 to 1 should usually suffice, with λ spacing values of of 0.05-0.1 being common. Note that this is a broad generalization, and in fact many molecules will require a great deal more finesse, such as those that interact very strongly with the surrounding environment through hydrogen bonding. In this case, λ spacing may need to be decreased, such that more points are used, perhaps even in an asymmetric fashion. For decoupling van der Waals interactions, λ spacing can be much trickier. For reasons described by Shirts et al. and elsewhere, a great deal of λ points may be necessary to properly describe the transformation. Clustering λ points in regions where the slope of the ∂H/∂λ curve is steep may be necessary. For the purposes of this tutorial, we will use equidistant λ spacing of 0.05, but in many cases, one may need to use different spacing, especially clustering values in the range of 0.6 ≤ λ ≤ 0.8. For each value of λ, a complete workflow (energy minimization, equilibration, and data collection) must be conducted. I generally find it most efficient to run these jobs as batches, passing the output of one step directly into the next. The workflow utilized here will be:
The .mdp files for the different steps of the tutorial (for λ = 0 only) are provided at the following links: I am also making available an accessory Perl script, write_mdp.pl, that you can use to very quickly produce all of the input files needed for running these simulations. 你还可以下载一个 shell 脚本(job.sh),用于根据我将描述的工作流程运行这些作业。 If you issue: 如果你执行: perl write_mdp.pl em_steep.mdp
You will receive files named em_steep_0.mdp, em_steep_1.mdp, ... em_steep_20.mdp, with the relevant values of init_lambda_state inserted into the filename. Analogously, you can produce the rest of your .mdp files in the same way (nvt.mdp, npt.mdp, and md.mdp). In job.sh, you will need to change the values of Notes on .mdp settings | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



|
| |
| free_energy = yes | Indicates that we are doing a free energy calculation, and that interpolation between the A and B states of the chosen molecule (defined below) will occur. 表示我们正在进行自由能计算,并且将在所选分子(定义如下)的 A 和 B 状态之间进行插值。 |
|
| |
| init_lambda_state = 0 | In previous GROMACS versions, the "init_lambda" keyword specified a single value of λ directly. Since version 5.0, λ is now a vector that allows for transformation of bonded and nonbonded interactions. With the init_lambda_state keyword, we specify an index (starting from zero) of the vector to be utilized in the simulation (more on this later). 在之前的 GROMACS 版本中,"init_lambda"关键字直接指定了一个λ的单一值。自 5.0 版本起,λ现在是一个向量,允许对键合和非键合相互作用进行转换。通过 init_lambda_state 关键字,我们指定在模拟中要使用的向量的索引(从零开始)(更多内容将在后面介绍)。 |
|
| |
| delta_lambda = 0 | The value of λ can be incremented by some amount per timestep (i.e., δλ/δt) in a technique called "slow growth." This method can have significant errors associated with it, and thus we will make no time-dependent changes to our λ values. λ的值可以通过每个时间步长增加一定的量(即δλ/δt)在一种称为"慢增长"的技术中实现。这种方法与其相关联的显著误差,因此我们将不对我们的λ值进行时间相关的改变。 |
|
| |
| fep_lambdas = (nothing) | You will note that this keyword is not specified. In previous GROMACS versions, the use of init_lambda and foreign_lambda controlled the value of λi and the additional values of λ for which energy differences would be evaluated for configurations at λi. This is no longer the case. One can explicitly set values of λ in the fep_lambdas keyword, but instead we allow the calc_lambda_neighbors keyword (see below) to automatically determine these additional values. 您会注意到这个关键字没有指定。在之前的 GROMACS 版本中,init_lambda 和 foreign_lambda 的使用控制了λ的值以及λ的附加值,这些附加值用于计算λ状态下配置的能量差。现在情况不再如此。用户可以在 fep_lambdas 关键字中显式设置λ的值,但我们允许 calc_lambda_neighbors 关键字(见下文)自动确定这些附加值。 |
|
| |
| calc_lambda_neighbors = 1 | The number of neighboring windows for which energy differences are computed with respect to λi. For instance, if init_lambda_state is set to 10, then energy differences with respect to λ states 9 and 11 are computed during the run with calc_lambda_neighbors = 1. 用于计算与λ相关的能量差的相邻窗口数量。例如,如果 init_lambda_state 设置为 10,那么在 calc_lambda_neighbors = 1 的情况下,会计算λ状态 9 和 11 的能量差。 |
|
| |
| vdw_lambdas = ... | An array of λ values for the transformation of van der Waals interactions. van der Waals 相互作用的λ值数组。 |
|
| |
| coul_lambdas = ... | An array of λ values for the transformation of Coulombic (electrostatic) interactions. Coulombic(静电)相互作用的λ值数组。 |
|
| |
| bonded_lambdas = ... | An array of λ values for the transformation of bonded interactions. 用于键合相互作用转换的λ值数组。 |
|
| |
| restraint_lambdas = ... | An array of λ values for the transformation of restraints. 用于约束转换的λ值数组。 |
|
| |
| mass_lambdas = ... | An array of λ values for the transformation of atomic masses; used if tranforming the chemical nature of the molecule. 用于原子质量转换的一系列λ值;如果转换分子的化学性质时使用。 |
|
| |
| temperature_lambdas = ... | An array of λ values for the transformation of temperatures; used only for simulated tempering. 用于温度转换的一系列λ值;仅用于模拟退火。 |
|
| |
| sc-alpha = 0.5 | The value of the α scaling factor used in the "soft-core" Lennard-Jones equation. See equations 4.124 - 4.126 in the GROMACS manual, section 4.5.1, for a complete description of this term, as well as the next three. α缩放因子在"软核"伦纳德-琼斯方程中的值。有关该术语以及接下来三个术语的完整描述,请参阅 GROMACS 手册第 4.5.1 节中的公式 4.124 至 4.126。 |
|
| |
| sc-coul = no | Transform Coulombic interactions linearly. This is the default behavior, but is written out for clarity. 线性转换库仑相互作用。这是默认行为,但为了清晰起见,这里明确写出。 |
|
| |
| sc-power = 1.0 | The power for λ in the soft-core equation. λ在软核方程中的幂。 |
|
| |
| sc-sigma = 0.3 | The value of σ assigned to any atom types that have C6 or C12 parameters equal to zero or σ < sc-sigma (typically H atoms). This value is used in the soft-core Lennard-Jones equation. 为任何具有 C6 或 C12 参数等于零或σ < sc-sigma 的原子类型分配的σ值(通常为 H 原子)。此值用于软核 Lennard-Jones 方程。 |
|
| |
| couple-moltype = Methane | The name of the [moleculetype] in that will have its topology interpolated from state A to state B. Note that the name given here must match a [moleculetype] name, and not the residue name. In some cases these may be the same, but I have maintained different names for the [moleculetype] and component residue for instructive purposes. 那个[moleculetype]的名称将从状态 A 插值到状态 B。请注意,这里提供的名称必须与[moleculetype]名称匹配,而不是残基名称。在某些情况下它们可能是相同的,但我为了说明目的,为[moleculetype]和组件残基保留了不同的名称。 |
|
| |
| couple-lambda0 = vdw | The types of nonbonded interactions that are present in state A between the interpolated [moleculetype] and the remainder of the system. The value "vdw" indicates that only van der Waals terms are active between methane and water; there are no solute-solvent Coulombic interactions. 在状态 A 中,插值[moleculetype]与系统其余部分之间的非键合相互作用类型。值"vdw"表示甲烷和水之间仅激活范德华项;没有溶质-溶剂库仑相互作用。 |
|
| |
| couple-lambda1 = none | The types of nonbonded interactions that are present in state B between the interpolated [moleculetype] and the remainder of the system. The value "none" indicates that both van der Waals and Coulombic interactions are off in state B. Relative to couple-lambda0, this indicates that only van der Waals terms have been turned off. 在状态 B 中,插值[moleculetype]与系统其余部分之间的非键相互作用类型。"none"值表示在状态 B 中范德华力和库仑力均关闭。相对于 couple-lambda0,这表示仅关闭了范德华力项。 |
|
| |
| couple-intramol = no | Do not decouple intramolecular interactions. That is, the λ factor is applied to only solute-solvent nonbonded interactions and not solute-solute nonbonded interactions. 不要解耦分子内相互作用。也就是说,λ因子仅应用于溶质-溶剂非键相互作用,而不应用于溶质-溶质非键相互作用。 Setting "couple-intramol = yes" is useful for larger molecules that may have intramolecular interactions occuring at longer distance (e.g. peptides or other polymers). Otherwise, distal parts of the molecule will interact via explicit pair interactions in an unnaturally strong manner (since solute-solvent interactions are weakened as a function of λ, but the intramolecular terms would not be), giving rise to configurations that will incorrectly bias the resulting free energy. 设置"couple-intramol = yes"对于可能存在较远距离分子内相互作用的大分子(例如肽或其他聚合物)很有用。否则,分子的远端部分将通过显式对相互作用以一种不自然的方式相互作用(因为溶质-溶剂相互作用随着λ的减弱而减弱,但分子内项不会减弱),从而产生会错误地偏置所得自由能的构型。 |
|
| |
| nstdhdl = 10 | The frequency with which ∂H/∂λ and ΔH are written to dhdl.xvg. A value of 100 would probably suffice, since the resulting values will be highly correlated and the files will get very large. You may wish to increase this value to 100 for your own work. 将 ∂H/∂λ 和 ΔH 写入 dhdl.xvg 的频率。100 的值可能就足够了,因为结果值会高度相关,而且文件会变得非常大。你可以根据自己的工作需要将这个值增加到 100。 |
|
| |
There are also several differences in the .mdp settings that will be used here relative to the settings used by Shirts et al.:
相对于 Shirts 等人使用的设置,这里在 .mdp 设置中也有几个不同之处:
- rlist=rcoulomb=rvdw=1.2. In order to use PME, rlist must be equal to rcoulomb, a check that was enforced in grompp sometime after the release of version 3.3.1. The Verlet cutoff scheme introduced in version 4.6 that allows for buffered neighbor lists also requires rvdw=rcoulomb. The value of rlist will be tuned during the run for the purposes of energy conservation, thus providing the necessary buffer for the switched van der Waals interactions. The switching range (from 1.0-1.2 nm) agrees with the methods of Shirts et al. for the treatment of solute-solvent interactions.
rlist=rcoulomb=rvdw=1.2。为了使用 PME,rlist 必须等于 rcoulomb,这是一个在 grompp 版本 3.3.1 发布后有时强制执行的检查。版本 4.6 中引入的允许使用缓冲邻居列表的 Verlet 截断方案也需要 rvdw=rcoulomb。运行期间会调整 rlist 的值以实现能量守恒,从而为切换的范德华相互作用提供必要的缓冲。切换范围(1.0-1.2 nm)与 Shirts 等人处理溶剂-溶质相互作用的方法一致。 - Temperature coupling is handled through the use of the Langevin integrator (the "sd" setting specifies "stochastic dynamics," e.g. with a random friction force), rather than an Andersen thermostat.
温度耦合通过使用朗之万积分器("sd"设置指定"随机动力学",例如使用随机摩擦力)来处理,而不是使用安德森恒温器。 - tau_t = 1.0. It is important to illustrate this particular value, because when using the Langevin integrator, this value corresponds to an inverse friction coefficient, therefore in ps-1. The value of 1.0 avoids over-damping the dynamics of water.
tau_t = 1.0。重要的是要说明这个特定值,因为在使用朗之万积分器时,这个值对应于一个倒数摩擦系数,因此以 ps -1 为单位。1.0 的值避免了过度阻尼水的动力学。
Let us take a moment to dissect the λ vectors a bit more closely. As an example, for init_lambda_state = 0, that means we are specifying that the state in the transformation corresponds to the vector with index 0 in the *_lambdas keywords, like so:
让我们更仔细地分析一下λ向量。例如,对于 init_lambda_state = 0,这意味着我们指定转换中的状态对应于*_lambdas 关键字中索引为 0 的向量,如下所示:
; init_lambda_state 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
vdw_lambdas = 0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40 0.45 0.50 0.55 0.60 0.65 0.70 0.75 0.80 0.85 0.90 0.95 1.00
coul_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
bonded_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
restraint_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
mass_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
temperature_lambdas = 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
Setting initial_lambda_state = 1 would correspond to the next column to the right (λ for van der Waals = 0.05), while initial_lambda_state = 20 would specify the final column (λ vector), in which the value of λ for van der Waals = 1.0. For the purposes of this tutorial, we are only transforming van der Waals interactions, leaving everything related to charges, bonded parameters, restraints, etc. alone. Please note that, in this example, where charges are zero in the topology and the .mdp settings never indicate that charges should be present (neither couple-lambda0 nor couple-lambda1 include 'q'), the choice of values for coul_lambdas is irrelevant, but for the sake of being pedantic, the .mdp files make clear that no charge transformation is taking place.
将 initial_lambda_state 设置为 1 对应右侧的下一列(van der Waals λ = 0.05),而 initial_lambda_state 设置为 20 则指定最后一列(λ 向量),其中 van der Waals λ 的值为 1.0。在本教程中,我们仅转换 van der Waals 相互作用,而电荷、键合参数、约束等均保持不变。请注意,在此示例中,拓扑中电荷为零,且 .mdp 设置从未指示应存在电荷(既 couple-lambda0 也 couple-lambda1 均不包含 'q'),因此 coul_lambdas 的值选择无关紧要,但为严谨起见,.mdp 文件明确说明未进行电荷转换。
Step Four: Energy Minimization
第四步:能量最小化
| The job.sh script I provide for running these calculations will create the following directory hierarchy: Lambda_0/
Lambda_0/EM/
Lambda_0/NVT/
Lambda_0/NPT/
Lambda_0/Production_MD/
This way, all steps in the workflow are executed within a single directory for each value of init_lambda_state. I find this to be a convenient way to organize the jobs and their output. The script also assumes that the .mdp files are also organized hierarchically within some directory $MDP/
$MDP/EM/
$MDP/NVT/
$MDP/NPT/
$MDP/Production_MD/
As described before, energy minimization will be conducted using the steepest descent method. The relevant section in the job.sh script is: mkdir Lambda_$LAMBDA
cd Lambda_$LAMBDA
#################################
# ENERGY MINIMIZATION 1: STEEP #
#################################
echo "Starting minimization for lambda = $LAMBDA..."
mkdir EM
cd EM
# Iterative calls to grompp and mdrun to run the simulations
gmx grompp -f $MDP/em_steep_$LAMBDA.mdp -c $FREE_ENERGY/methane_water.gro
-p $FREE_ENERGY/topol.top -o min$LAMBDA.tpr
gmx mdrun -deffnm min$LAMBDA
echo "Minimization complete." |
Step Five: Equilibration 第五步:平衡
| The system will be equilibrated in two phases, the first at constant volume (NVT), the second at constant pressure (NPT). #####################
# NVT EQUILIBRATION #
#####################
echo "Starting constant volume equilibration..."
cd ../
mkdir NVT
cd NVT
gmx grompp -f $MDP/nvt_$LAMBDA.mdp -c ../EM/min$LAMBDA.gro
-p $FREE_ENERGY/topol.top -o nvt$LAMBDA.tpr
gmx mdrun -deffnm nvt$LAMBDA
echo "Constant volume equilibration complete."
#####################
# NPT EQUILIBRATION #
#####################
echo "Starting constant pressure equilibration..."
cd ../
mkdir NPT
cd NPT
gmx grompp -f $MDP/npt_$LAMBDA.mdp -c ../NVT/nvt$LAMBDA.gro
-p $FREE_ENERGY/topol.top -t ../NVT/nvt$LAMBDA.cpt -o npt$LAMBDA.tpr
gmx mdrun -deffnm npt$LAMBDA
echo "Constant pressure equilibration complete." |
Step Six: Production MD 第六步:生产 MD
| Now that the system is equilibrated, we can begin production MD simulations, during which time we will collect ∂H/∂λ data. #################
# PRODUCTION MD #
#################
echo "Starting production MD simulation..."
cd ../
mkdir Production_MD
cd Production_MD
gmx grompp -f $MDP/md_$LAMBDA.mdp -c ../NPT/npt$LAMBDA.gro
-p $FREE_ENERGY/topol.top -t ../NPT/npt$LAMBDA.cpt -o md$LAMBDA.tpr
gmx mdrun -deffnm md$LAMBDA
echo "Production MD complete."
The whole workflow should take about 2.5 hours to complete on a GPU workstation. Since there are many simulations to run (21 sets of λ vectors), it is best to run these jobs on clusters where more processors are available such that jobs can be run simultaneously. Once all of the production simulations are complete, we can analyze the resulting data. |
Step Seven: Analysis 第七步:分析
| The bar module of GROMACS makes the calculation of ΔGAB very simple. Simply collect all of the md*.xvg files that were produced by mdrun (one for each value of λ) in the working directory and run gmx bar: gmx bar -f md*.xvg -o -oi
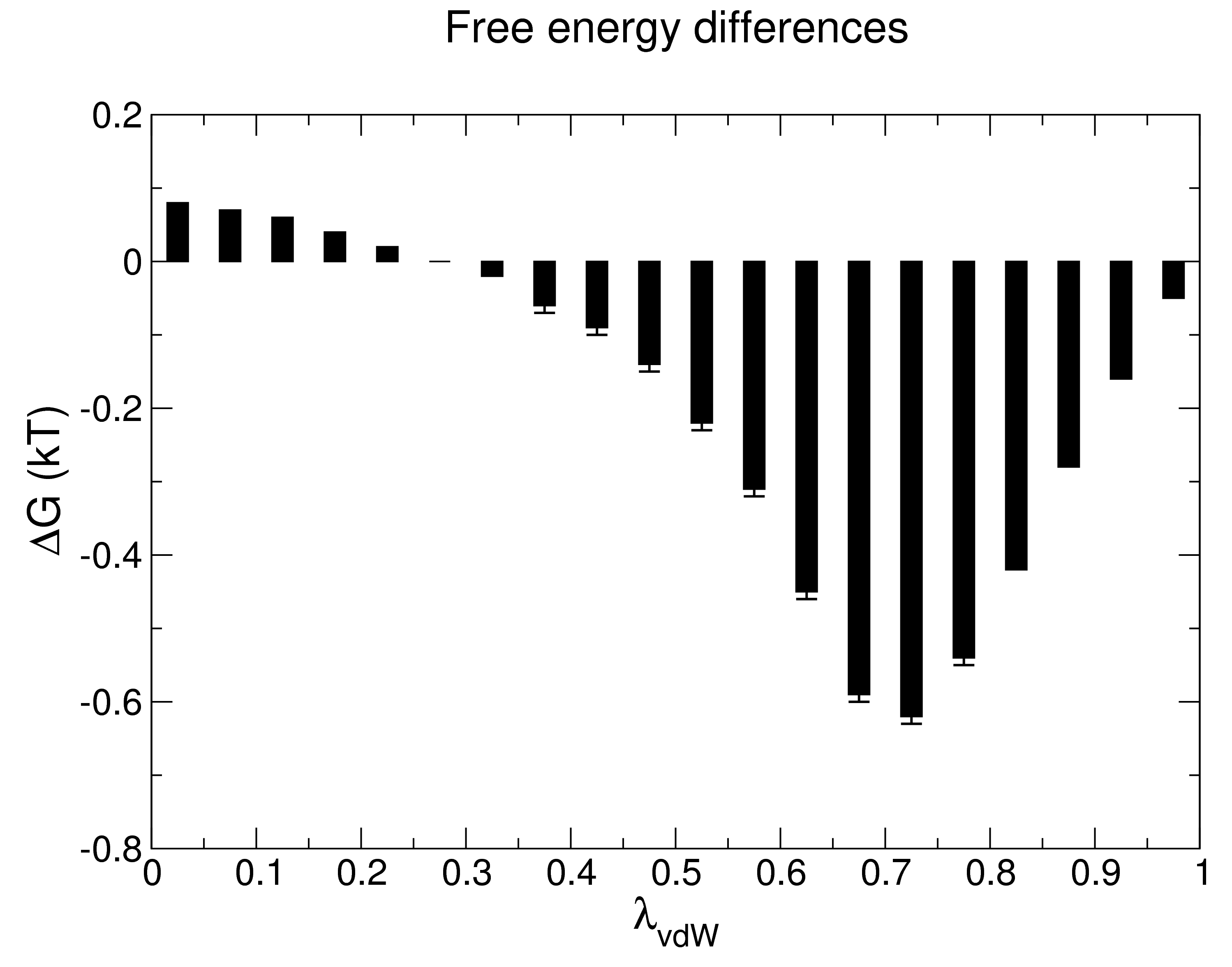
The program will print lots of useful information to the screen, in addition to producing three output files: bar.xvg, barint.xvg, and histogram.xvg. The screen output from gmx bar should look something like: Detailed results in kT (see help for explanation):
lam_A lam_B DG +/- s_A +/- s_B +/- stdev +/-
0 1 0.08 0.00 0.03 0.00 0.03 0.00 0.25 0.00
1 2 0.07 0.00 0.03 0.00 0.03 0.00 0.25 0.00
2 3 0.06 0.00 0.03 0.00 0.04 0.00 0.26 0.00
3 4 0.04 0.00 0.04 0.00 0.05 0.00 0.28 0.00
4 5 0.02 0.00 0.03 0.00 0.04 0.00 0.29 0.00
5 6 0.00 0.00 0.05 0.00 0.06 0.00 0.30 0.00
6 7 -0.02 0.00 0.05 0.00 0.06 0.00 0.32 0.00
7 8 -0.06 0.01 0.06 0.00 0.07 0.00 0.35 0.01
8 9 -0.09 0.01 0.06 0.00 0.07 0.00 0.37 0.00
9 10 -0.14 0.01 0.08 0.00 0.10 0.01 0.41 0.01
10 11 -0.22 0.01 0.09 0.01 0.12 0.01 0.46 0.00
11 12 -0.31 0.01 0.12 0.01 0.15 0.01 0.51 0.01
12 13 -0.45 0.01 0.17 0.01 0.20 0.01 0.57 0.00
13 14 -0.59 0.01 0.16 0.02 0.17 0.02 0.58 0.01
14 15 -0.62 0.01 0.11 0.01 0.11 0.01 0.47 0.00
15 16 -0.54 0.01 0.06 0.01 0.06 0.01 0.35 0.00
16 17 -0.42 0.00 0.03 0.01 0.03 0.01 0.25 0.00
17 18 -0.28 0.00 0.02 0.00 0.02 0.00 0.18 0.00
18 19 -0.16 0.00 0.01 0.00 0.01 0.00 0.13 0.00
19 20 -0.05 0.00 0.00 0.00 0.00 0.00 0.10 0.00
Final results in kJ/mol:
point 0 - 1, DG 0.19 +/- 0.00
point 1 - 2, DG 0.17 +/- 0.01
point 2 - 3, DG 0.14 +/- 0.00
point 3 - 4, DG 0.09 +/- 0.01
point 4 - 5, DG 0.06 +/- 0.00
point 5 - 6, DG 0.01 +/- 0.01
point 6 - 7, DG -0.06 +/- 0.01
point 7 - 8, DG -0.14 +/- 0.02
point 8 - 9, DG -0.23 +/- 0.01
point 9 - 10, DG -0.35 +/- 0.01
point 10 - 11, DG -0.53 +/- 0.01
point 11 - 12, DG -0.77 +/- 0.03
point 12 - 13, DG -1.12 +/- 0.03
point 13 - 14, DG -1.46 +/- 0.02
point 14 - 15, DG -1.53 +/- 0.04
point 15 - 16, DG -1.35 +/- 0.02
point 16 - 17, DG -1.04 +/- 0.01
point 17 - 18, DG -0.70 +/- 0.01
point 18 - 19, DG -0.40 +/- 0.00
point 19 - 20, DG -0.13 +/- 0.00
total 0 - 20, DG -9.13 +/- 0.09
The value of ΔGAB I obtained is -9.13 ± 0.09 kJ mol-1, or -2.18 ± 0.02 kcal mol-1. Since the process I conducted for this demonstration was the decoupling of methane, the reverse process (the introduction of uncharged methane into water, thus the actual hydration energy of the process) corresponds to a ΔGAB of 2.18 ± 0.02 kcal mol-1 (assuming reversibility), in good agreement with the value obtained by Shirts et al. of 2.24 ± 0.01 kcal mol-1 (per Table II of the Supporting Information of that paper), despite the fact that I used about half the number of λ vectors for the van der Waals transformation than the original authors did, in addition to the other changes described previously. Technically, this is all we need to do to arrive at our answer, but gmx bar also prints a number of useful output files (all of which are optional). Their contents are worth exploring here, as they provide a great level of detail into the decoupling process and the success of sampling. Output file 1: bar.xvg |
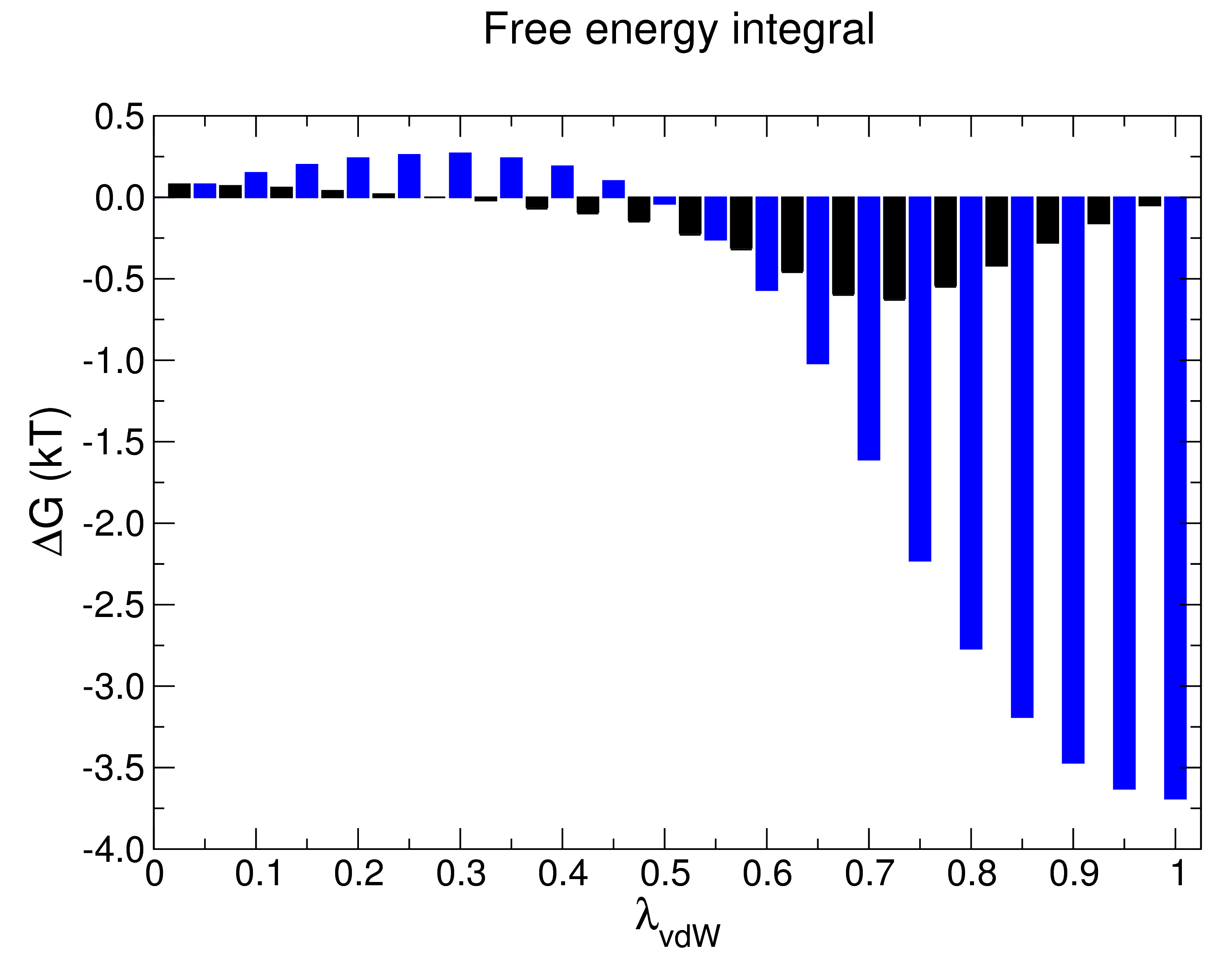
Advanced Applications 高级应用
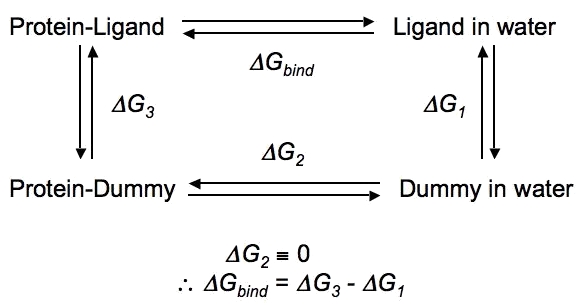
| One common application of calculating free energies is to determine the of ΔG of binding between a ligand and a receptor (e.g., a protein), you would need to perform (de)coupling of the ligand in complex with the receptor and in bulk solution, since ΔG is (in this case) the sum of the free energy change of complexation of the ligand and receptor (i.e. the introduction of the ligand into the binding site) and the free energy of desolvating the ligand (i.e. removing it from solution): ΔGbind = ΔGcomplexation + ΔGdesolvation ΔGsolvation = -ΔGdesolvation ∴ ΔGbind = ΔGcomplexation - ΔGsolvation ΔG of binding will be the difference between these two states, and can be calculated through the use of the following (somewhat simplified) thermodynamic cycle, wherein ΔG1 is ΔGsolvation and ΔG3 is ΔGcomplexation in the equations above.
A particularly detailed thermodynamic cycle for this type of process can be found in Figure 1 of this paper. The transformation of a fully-interacting species (e.g., the ligand) into a "dummy" (a set of atomic centers in the configuration of the ligand that does not engage in any nonbonded interactions with its surroundings) requires turning off both van der Waals interactions (as demonstrated in this tutorial) and Coulombic interactions between the solute of interest and its surrounding environment. The complete series of transformations for methane is shown below:
In the above thermodynamic cycle, ΔG1 and ΔG3 actually represent the introduction of the ligand (starting as a dummy molecule), i.e. coupling rather than decoupling. The thermodynamic cycle above is extremely basic, and does not account for considerations like orientational restraints, conformational changes of the protein, etc. None of this literature will be discussed here. For most small molecules (generically named "LIG" below), the following settings might seem attractive: couple-moltype = LIG
couple-intramol = no
couple-lambda0 = none
couple-lambda1 = vdw-q
Special care must be taken in this case. In previous versions of GROMACS, such an approach was likely to be very unstable because removal of van der Waals terms while a system retains some charge allows oppositely-charged atoms to interact very closely (in the absence of the full effects of an electron cloud). The result would be unstable configurations and unreliable energies, even if the systems didn't collapse. Thus, it is more sound to approach the (de)coupling sequentially. In version 5.0, this is very easily done with the new λ vector capabilities: couple-moltype = LIG
couple-intramol = no
couple-lambda0 = none
couple-lambda1 = vdw-q
init_lambda_state = 0
calc_lambda_neighbors = 1
vdw_lambdas = 0.00 0.05 0.10 ... 1.00 1.00 1.00 ... 1.00
coul_lambdas = 0.00 0.00 0.00 ... 0.00 0.05 0.10 ... 1.00
In this case, the λ value for Coulombic interactions is always zero while the λ value for transforming van der Waals interactions changes. Then, the van der Waals interactions are fully on (λ = 1.00) while the Coulombic interactions are gradually turned on. Keeping track of the states to which λ = 0 and λ = 1 correspond is key to this process. In the above case, couple-lambda0 says interactions are off, while couple-lambda1 means interactions are on. These types of transformations can be useful for calculating hydration/solvation free energies, since this quantity is often used as target data in force field parametrization. |

Summary 摘要
| You have now calculated the free energy change for a simple transformation that has previously been calculated with high precision, the decoupling of van der Waals interactions between a simple solute (uncharged methane) and solvent (water). Hopefully this tutorial will provide you with the understanding necessary to take on more complex systems. If you have suggestions for improving this tutorial, if you notice a mistake, or if anything is otherwise unclear, please feel free to email me. Please note: this is not an invitation to email me for GROMACS problems. I do not advertise myself as a private tutor or personal help service. That's what the GROMACS User Forum is for. I may help you there, but only in the context of providing service to the community as a whole, not just the end user. Happy simulating! 模拟愉快! |



















 3386
3386

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











