






一、写在前面
目前单细胞已经走向多组学发展道路,不仅局限于mRNA,染色质可及性、DNA甲基化、蛋白质定量都开始上桌竞争。相较于建库测序技术的进步,算法与软件的更新总是慢上一排,即使是这样,目前也有超过数千种的单细胞测序相关算法。各项算法工具的推出也让各位产生了一定的选择困难,如何用稳定的、公平的、没有人工/批次效应的数据与方法测评这些软件工具也是一个难点。公平的基准测试要求计算机数据包含真实数据并模拟真实数据,因此需要真实的模拟器和基准测试研究。加州大学洛杉矶分校的团队就推出了一个统计模拟器 scDesign3,能够生成逼真的合成数据,包括细胞潜在结构、细胞轨迹、组学特征、空间位置、实验设计、技术平台等信息。scDesign3能够模拟10X Visium、Slide-seq、10x scATAC-seq、sci-ATAC-seq多种单细胞/空间技术(Figure 1)。我们之前提到的scRNA-Seq双细胞过滤手册,其中也用到了scDesign生成的模拟数据。
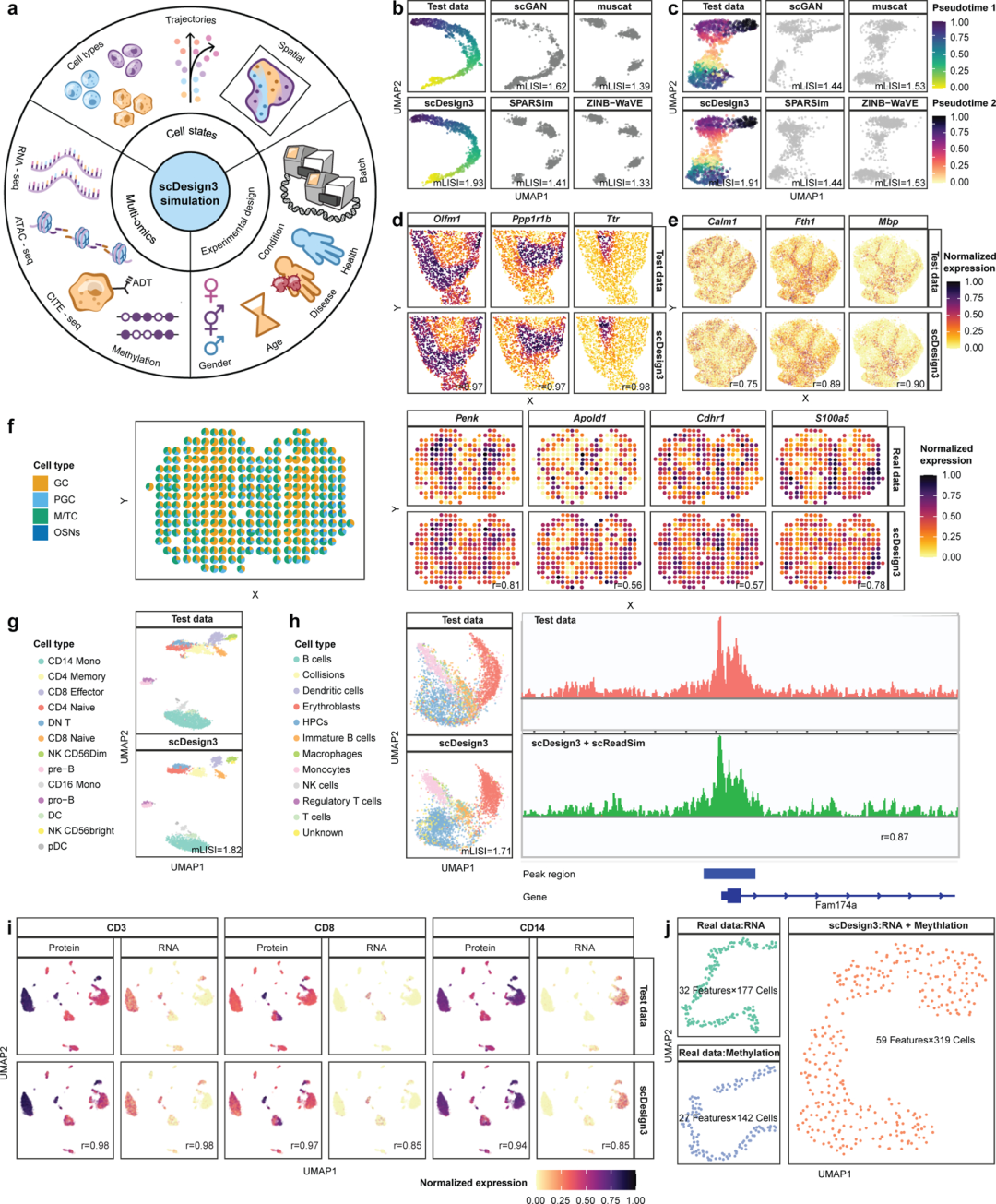
scDesign3 生成各种单细胞和空间组学技术的真实合成数据
二、演示
if(!require(scDesign3))devtools::install_github("SONGDONGYUAN1994/scDesign3")## Loading required package: scDesign3## Registered S3 method overwritten by 'scDesign3':
## method from
## predict.gamlss gamlssif(!require(scDesign3))BiocManager::install('SingleCellExperiment')
if(!require(ggplot2))install.packages('ggplot2')## Loading required package: ggplot2if(!require(ExperimentHub))BiocManager::install("ExperimentHub")## Loading required package: ExperimentHub## Loading required package: BiocGenerics##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
## match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
## Position, rank, rbind, Reduce, rownames, sapply, setdiff, sort,
## table, tapply, union, unique, unsplit, which.max, which.min## Loading required package: AnnotationHub## Loading required package: BiocFileCache## Loading required package: dbplyr## Registered S3 method overwritten by 'bit':
## method from
## print.ri gamlssif(!require(DuoClustering2018))BiocManager::install('DuoClustering2018')## Loading required package: DuoClustering2018## snapshotDate(): 2023-10-24if(!require(scran))BiocManager::install('scran')## Loading required package: scran## Loading required package: SingleCellExperiment## Loading required package: SummarizedExperiment## Loading required package: MatrixGenerics## Loading required package: matrixStats##
## Attaching package: 'MatrixGenerics'## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars## Loading required package: GenomicRanges## Loading required package: stats4## Loading required package: S4Vectors##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unname## Loading required package: IRanges## Loading required package: GenomeInfoDb## Loading required package: Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## Attaching package: 'Biobase'## The following object is masked from 'package:MatrixGenerics':
##
## rowMedians## The following objects are masked from 'package:matrixStats':
##
## anyMissing, rowMedians## The following object is masked from 'package:ExperimentHub':
##
## cache## The following object is masked from 'package:AnnotationHub':
##
## cache## Loading required package: scuttleif(!require(tidyverse))install.packages('tidyverse')## Loading required package: tidyverse## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──## ✔ tibble 3.2.1 ✔ dplyr 1.1.3
## ✔ tidyr 1.3.0 ✔ stringr 1.5.0
## ✔ readr 2.1.4 ✔ forcats 0.5.1
## ✔ purrr 1.0.2## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::collapse() masks IRanges::collapse()
## ✖ dplyr::combine() masks Biobase::combine(), BiocGenerics::combine()
## ✖ dplyr::count() masks matrixStats::count()
## ✖ dplyr::desc() masks IRanges::desc()
## ✖ tidyr::expand() masks S4Vectors::expand()
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::first() masks S4Vectors::first()
## ✖ dplyr::ident() masks dbplyr::ident()
## ✖ dplyr::lag() masks stats::lag()
## ✖ BiocGenerics::Position() masks ggplot2::Position(), base::Position()
## ✖ purrr::reduce() masks GenomicRanges::reduce(), IRanges::reduce()
## ✖ dplyr::rename() masks S4Vectors::rename()
## ✖ dplyr::slice() masks IRanges::slice()
## ✖ dplyr::sql() masks dbplyr::sql()theme_set(theme_bw())# 生成包含不同细胞比例的数据:
# 读取SingleCellExperiment测试数据:
sce <- get("sce_filteredExpr10_Zhengmix4eq")(metadata = FALSE)## see ?DuoClustering2018 and browseVignettes('DuoClustering2018') for documentation## loading from cache# 将细胞类型整理为因子变量:
colData(sce)$cell_type = as.factor(colData(sce)$phenoid)
# 只保留200个高变基因,加快后续运行速度:
ngene <- 200
# log1p标准化:
logcounts(sce) <- log1p(counts(sce))
# 识别高变基因:
temp_sce <- modelGeneVar(sce)
# 取出高变基因:
chosen <- getTopHVGs(temp_sce, n = ngene)
# 仅保留高变基因数据:
sce <- sce[chosen,]
##### 模拟一个与原数据细胞类型一致的新数据 ######
# 设置随机数,保证结果可重复性:
set.seed(123)
example_simu <- scdesign3(
sce = sce,
assay_use = "counts", # 参考矩阵
celltype = "cell_type",# 参考的分类变量
pseudotime = NULL,# 拟时间的变量名,没有提供就写NULL
spatial = NULL,# 空间坐标信息,分别提供x,y的坐标向量变量,没有提供就写NULL
other_covariates = NULL,# 其它参与的协变量变量名
mu_formula = "cell_type",# 模型期望值
sigma_formula = "cell_type",# 模型方差
family_use = "nb",# 指定用于模拟的统计分布家族,这里使用 "nb" 表示负二项分布(用于counts)。
n_cores = 2,# 并行计算的核心数
usebam = FALSE,# 是否使用bam文件进行加速模拟
corr_formula = "cell_type",# 模拟的相关性结构变量名
copula = "gaussian",# 'gaussian'或vine'任选一个
DT = TRUE,# 是否将discrete data转换为continuous
pseudo_obs = FALSE,# 如果是T,经验分位数而不是理论分位数来拟合copula
return_model = FALSE,# 是否返回模型
nonzerovar = FALSE,# 如果为T,则小于0的值会被归零
parallelization = "pbmcmapply"# 并行策略,在'mcmapply', 'bpmapply', 'pbmcmapply'中选择
)## Input Data Construction Start## Input Data Construction End## Start Marginal Fitting## Marginal Fitting End## Start Copula Fitting## Convert Residuals to Multivariate Gaussian## Converting End## Copula group b.cells starts## Copula group naive.cytotoxic starts## Copula group cd14.monocytes starts## Copula group regulatory.t starts## Copula Fitting End## Start Parameter Extraction## Parameter
## Extraction End## Start Generate New Data## Use Copula to sample a multivariate quantile matrix## Sample Copula group b.cells starts## Sample Copula group naive.cytotoxic starts## Sample Copula group cd14.monocytes starts## Sample Copula group regulatory.t starts## New Data Generating End# 结果是一个包含count、协变量、模型参数的list:
names(example_simu)## [1] "new_count" "new_covariate" "model_aic" "model_bic"
## [5] "marginal_list" "corr_list"# 构建新的sce对象:
simu_sce <- SingleCellExperiment(list(counts = example_simu$new_count),
colData = example_simu$new_covariate)
logcounts(simu_sce) <- log1p(counts(simu_sce))
# 模拟数据出炉
simu_sce## class: SingleCellExperiment
## dim: 200 3555
## metadata(0):
## assays(2): counts logcounts
## rownames(200): ENSG00000019582 ENSG00000204287 ... ENSG00000177156
## ENSG00000101220
## rowData names(0):
## colnames(3555): b.cells6276 b.cells6144 ... regulatory.t1084
## regulatory.t9696
## colData names(1): cell_type
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):# 看一下模拟数据的降维:
plot_reduceddim(ref_sce = sce,
sce_list = list(simu_sce),
name_vec = c("Reference",
"simulated sce"),
assay_use = "logcounts",
if_plot = TRUE,
color_by = "cell_type",
n_pc = 20)## 'as(<dgTMatrix>, "dgCMatrix")' is deprecated.
## Use 'as(., "CsparseMatrix")' instead.
## See help("Deprecated") and help("Matrix-deprecated").## $p_pca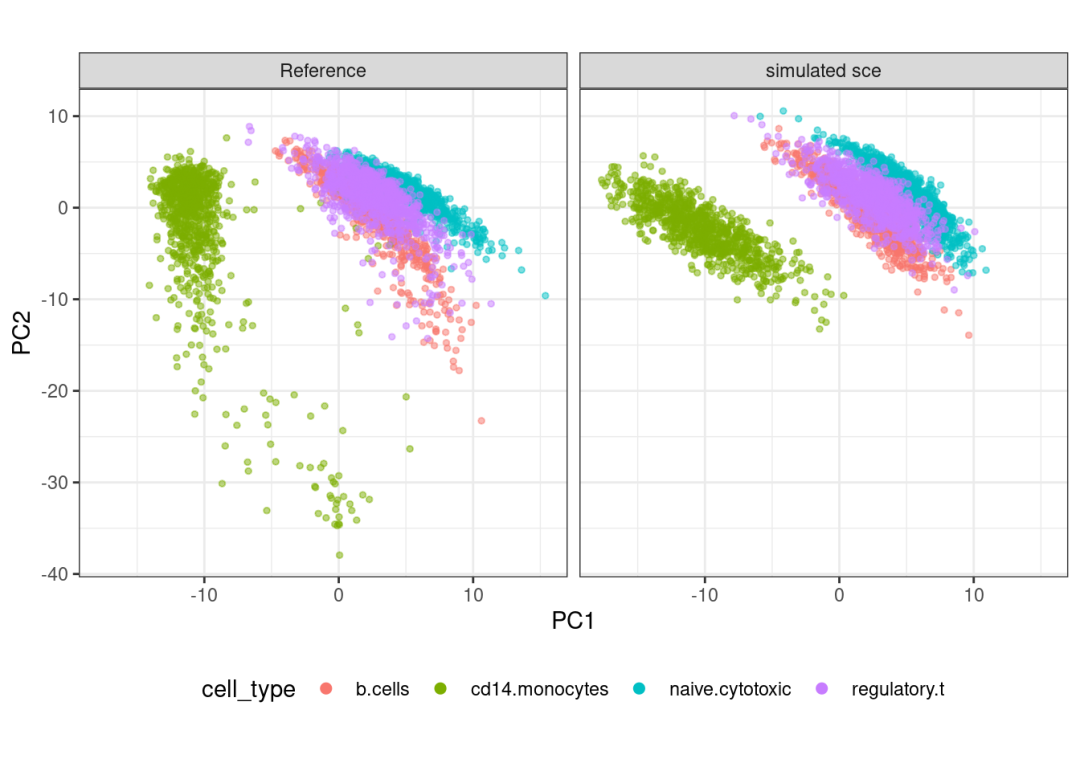
### 环境信息 ###sessionInfo()# R version 4.3.1 (2023-06-16)# Platform: x86_64-pc-linux-gnu (64-bit)# Running under: Ubuntu 20.04.6 LTS# Matrix products: default# BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3# LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0# locale:# [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8# [6] LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C# [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C# time zone: Etc/UTC# tzcode source: system (glibc)# attached base packages:# [1] stats4 stats graphics grDevices utils datasets methods base# other attached packages:# [1] DuoClustering2018_1.20.0 ExperimentHub_2.4.0 AnnotationHub_3.4.0 BiocFileCache_2.8.0 dbplyr_2.3.4# [6] forcats_0.5.1 stringr_1.5.0 dplyr_1.1.3 purrr_1.0.2 tidyr_1.3.0# [11] tibble_3.2.1 tidyverse_1.3.1 scran_1.24.0 scuttle_1.6.2 ggplot2_3.3.6# [16] SingleCellExperiment_1.22.0 SummarizedExperiment_1.30.2 GenomicRanges_1.52.0 GenomeInfoDb_1.36.4 IRanges_2.34.1# [21] S4Vectors_0.38.2 MatrixGenerics_1.12.3 matrixStats_1.0.0 scDesign3_1.1.3 readr_2.1.4# [26] shinyjs_2.1.0 shiny_1.7.5 sendmailR_1.4-0 future_1.33.0 SeuratObject_4.1.4# [31] Seurat_4.4.0 bigmemory_4.6.1 Biobase_2.60.0 BiocGenerics_0.46.0# loaded via a namespace (and not attached):# [1] vroom_1.6.4 goftest_1.2-3 gamlss_5.4-3 Biostrings_2.68.1# [5] vctrs_0.6.3 spatstat.random_3.1-6 digest_0.6.33 png_0.1-8# [9] shape_1.4.6 registry_0.5-1 ggrepel_0.9.3 deldir_1.0-9# [13] parallelly_1.36.0 MASS_7.3-60 reprex_2.0.1 reshape2_1.4.4# [17] httpuv_1.6.11 foreach_1.5.2 withr_2.5.1 ggrastr_1.0.1# [21] xfun_0.40 ggpubr_0.4.0 ellipsis_0.3.2 survival_3.5-7# [25] memoise_2.0.1 ggbeeswarm_0.6.0 ggsci_2.9 systemfonts_1.0.4# [29] zoo_1.8-12 GlobalOptions_0.1.2 pbapply_1.7-2 prettyunits_1.2.0# [33] KEGGREST_1.40.1 promises_1.2.1 httr_1.4.7 rstatix_0.7.0# [37] globals_0.16.2 fitdistrplus_1.1-11
更多模拟数据生成可参考:
https://songdongyuan1994.github.io/scDesign3/docs/articles/scDesign3-cellType-vignette.html
参考文献:Song D, Wang Q, Yan G, Liu T, Sun T, Li JJ. scDesign3 generates realistic in silico data for multimodal single-cell and spatial omics. Nat Biotechnol. 2024 Feb;42(2):247-252.


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









