






节点负荷查看
我们一致致力于给大家提提供高性价比的计算设备:足够支持你完成硕博生涯的生信环境。大家在使用西柚云服务器时不知道有没有发现,有的时候计算的负荷很高,从而会使计算速度变慢。对于共享资源的合理分配,我们早就做了很多硬件/软件的准备。这时大家可以看一下自己的CPU负载是否过高(内存是你独享的,占用多少应该你已经知道啦)。如果你对下面的教程比较迷茫,可以先行学习:十小时学会Linux、生信Linux及服务器使用技巧、5.5h入门R语言
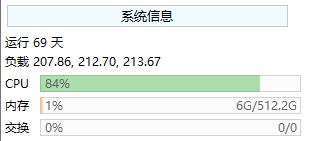
FinalShell的左上角就有系统信息可以直接查看:
当然,你也可以使用htop命令来查看CPU占用情况:
htop
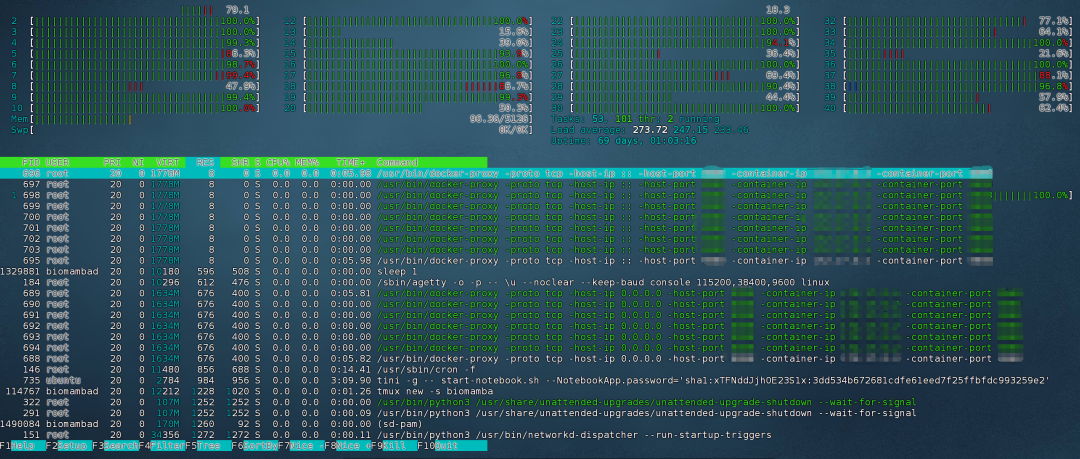
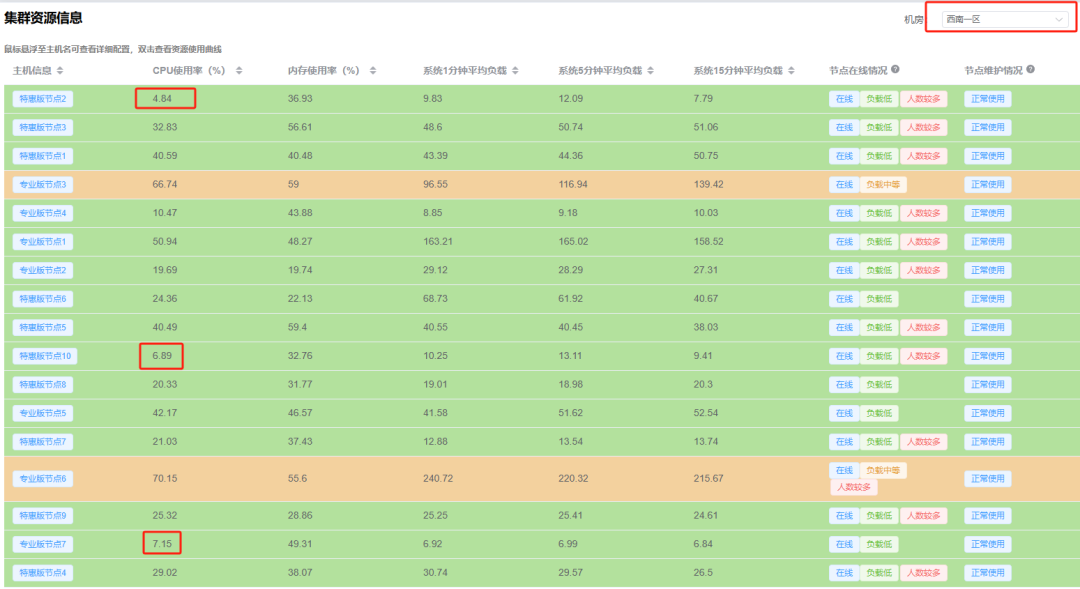
显然,在示例中,当前节点的CPU占用率高达84%(其实占用率可以超过100%)。长期的高占用这显然会影响我们的计算速度、甚至还有可能导致CPU硬件负荷高从而导致过热。好在我们给大家提供了数十个计算节点,并且按照用户数量定期添加算力。这时我们就可以来到大禹系统的控制台中的“仪表盘”。查看空闲计算节点,可以看到,我们的节点一篇绿色,很多节点的资源占用率只有个位数:
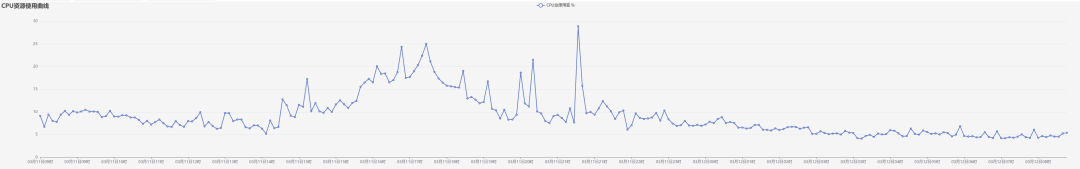
甚至还可以看到当前节点的CPU占用率随时间波动的曲线:
切换节点
这时大家可以进入实例列表:
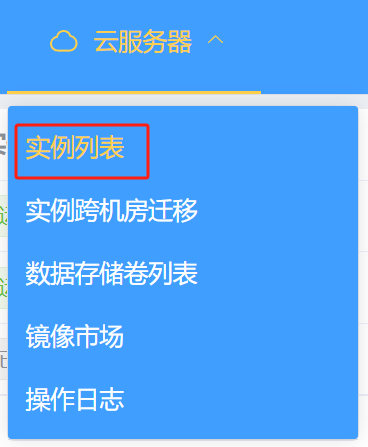
查看实例并进入详情界面:

需要先暂停你的实例运行,才可以完成迁移过程:
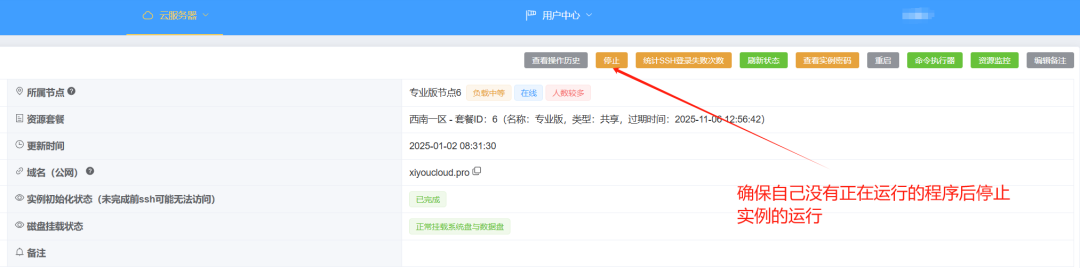
点击确认停止:
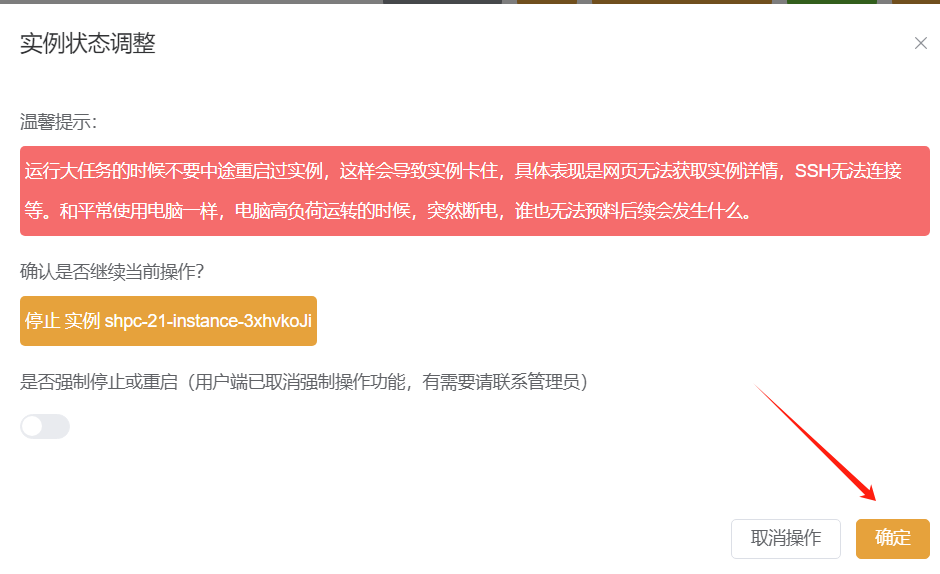
稍等片刻,确认实例停止后在实例列表中点击迁移:
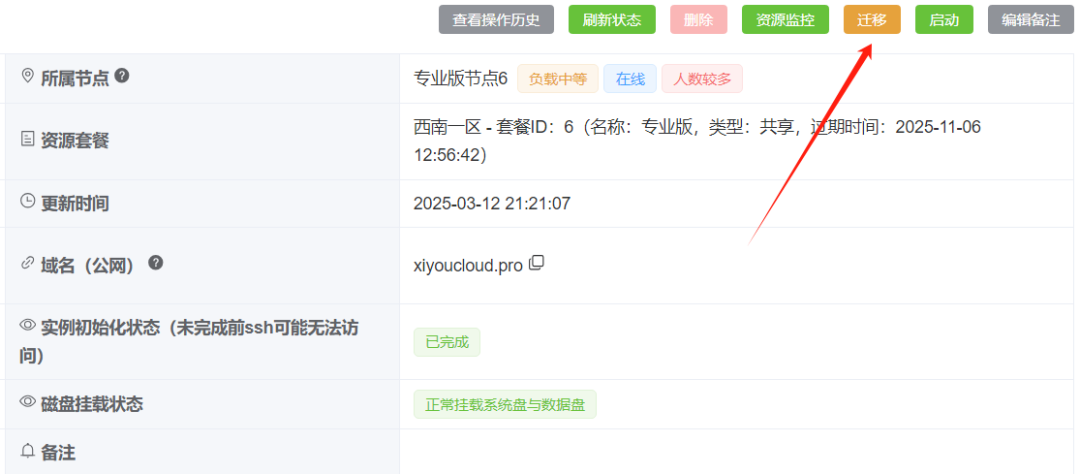
选择占用率低的节点:
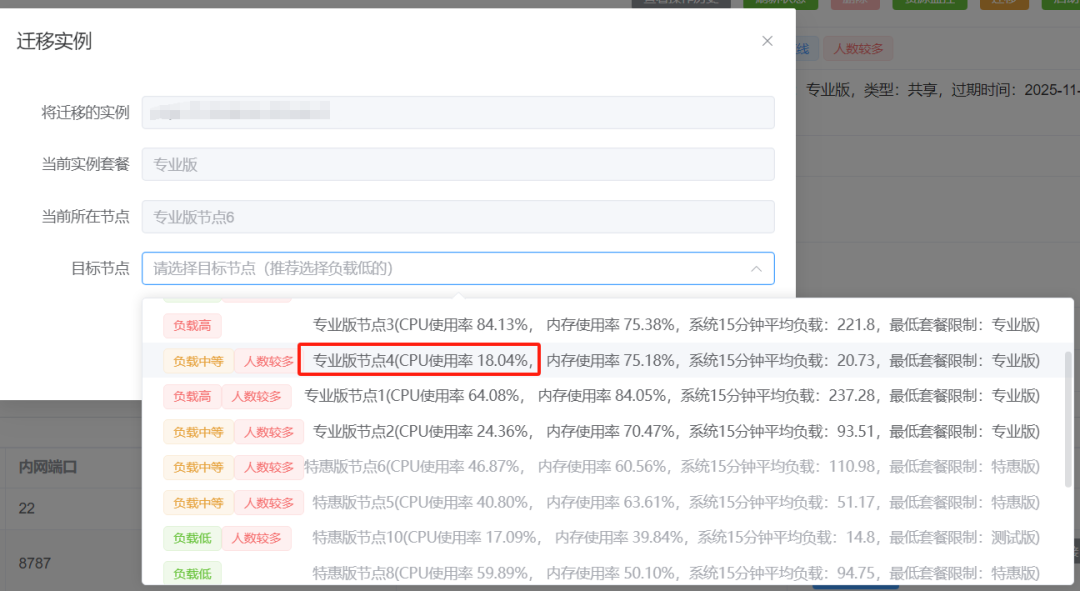
迁移完毕后记得重新启动实例:

重新启动后,无需更换原来的ssh与Rstudio-server访问方式,可以直接丝滑的接着使用:
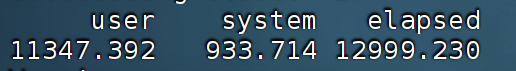
切换节点前:
切换节点后:
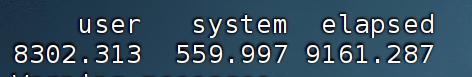
总时间大概快了1/4
测试代码与数据
顺序计算
# 测试代码system.time({library(Seurat)iri <- readRDS('./iri.intergrate.rds')iri[["percent.mt"]] <- PercentageFeatureSet(iri, pattern = "^MT-") #通过线粒体的序列数来对数据进行计算head(iri@meta.data, 5) #QC的数据存在meta.data里,可以用这个来查看前5行VlnPlot(iri, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)#用小提琴图来展示QC的结果,展示了每个barcode中基因的数目、UMI数目以及线粒体基因含量的分布情况plot1 <- FeatureScatter(iri, feature1 = "nCount_RNA", feature2 = "percent.mt")plot2 <- FeatureScatter(iri, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")CombinePlots(plots = list(plot1, plot2)) #高变基因,判断趋势及占比#质控,选子集,RNA数量在200与2500之间的,多的可能是低质量细胞或空drouplets,gene count多的可能是doublets or multipletsiri <- NormalizeData(iri, normalization.method = "LogNormalize", scale.factor = 10000)#用LogNormalize法对数据进行标准化(乘10000再取对数)数据存在iri[["RNA"]]@data.里#默认的方法也完成了log1p的操作,得到的结果就类似于TPM的对数iri <- FindVariableFeatures(iri, selection.method = "vst", nfeatures = 2000)# 筛选高变基因(输出2000个),用于下游的PCA及分群top10 <- head(VariableFeatures(iri), 10) #输出差异最大的十个基因plot1 <- VariableFeaturePlot(iri)plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)CombinePlots(plots = list(plot1, plot2)) #输出差异基因散点图(有无标签)iri <- ScaleData(iri, features = rownames(iri)) #将数据进行标准化,为后续的PCA分析做准备,数据存在iri[["RNA"]]@scale.data##for PCA DoHeatmap#iri <- ScaleData(iri)#only4VariableFeaturesiri <- ScaleData(iri, vars.to.regress = "percent.mt") #剔除不想要的变量(如线粒体的比例)#iri[["RNA"]]@scale.datairi <- RunPCA(iri, features = VariableFeatures(object = iri)) #PCA降维分析print(iri[["pca"]], dims = 1:5, nfeatures = 5) #打印PCA部分结果VizDimLoadings(iri, dims = 1:2, reduction = "pca")DimHeatmap(iri, dims = 1, cells = 500, balanced = TRUE) #三种PCA展示方式DimHeatmap(iri, dims = 1:15, cells = 500, balanced = TRUE) #展示15种PCA#筛选合适的维度:iri <- JackStraw(iri, num.replicate = 100) #重复计算次数iri <- ScoreJackStraw(iri, dims = 1:20) #计算维度这一步花的时间比较久JackStrawPlot(iri, dims = 1:15) #画出1到15个维度#JackStrawPlot相当于高级PCA,为挑选合适维度进行下游可视化提供依据ElbowPlot(iri) #利用ElbowPlot来评价PC,PCA切记为了结果好看而降低PC数iri <- FindNeighbors(iri, dims = 1:10)iri <- FindClusters(iri, resolution = 0.5)#细胞分群,resolution分辨率在细胞数在3000附近时一般设为0.4-1.2,resolution越大得到的类群越多head(Idents(iri), 5) #看前五个分类群ID#非线性降维方法:UMAP、tSNEiri <- RunUMAP(iri, dims = 1:10) #运行UMAP算法#sce.all <- RunUMAP(sce.all, dims = 1:30, min.dist = 0.01, n.neighbors = 4L)#可以调整亚群之间的距离iri <- RunTSNE(iri, dims = 1:10) #运行TSNE算法,TSNE算法运行时间较UMAP更久DimPlot(iri, reduction = "umap", label = TRUE)iri.markers <- FindAllMarkers(iri, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)})
设置并行计算在上面的实际计算代码前设置如下命令:
makecore <- function(workcore,memory){if(!require(Seurat))install.packages('Seurat')if(!require(future))install.packages('future')plan("multisession", workers = workcore)options(future.globals.maxSize= memory*1024*1024**2)}makecore(4,10)#这里以四线程,10GB为例
测试文件:(点击了解更多)


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









