







reading notes of《Artificial Intelligence in Drug Design》
文章目录
1.Introduction
- Methods that make use of a three-dimensional (3D) structure of the biological target of interest, typically a protein or a nucleic acid, are grouped under the term of structure-based drug design (SBDD).
- SBDD aims to predict compound that make optimal interactions in the target binding site.
- SBVS aims at processing in a timely manner large libraries of compounds. Small molecules are docked and ranked according to their estimated binding affinity. DNNs have been applied in structure-based virtual screening (SBVS) protocols. The role of DNN in this context is to make predictions that allow focusing docking calculations on the most promising compounds.
2. Scoring Functions
- Scoring functions are applied in molecular docking to predict binding modes and to estimate binding affinity of ligands to macromolecular targets.
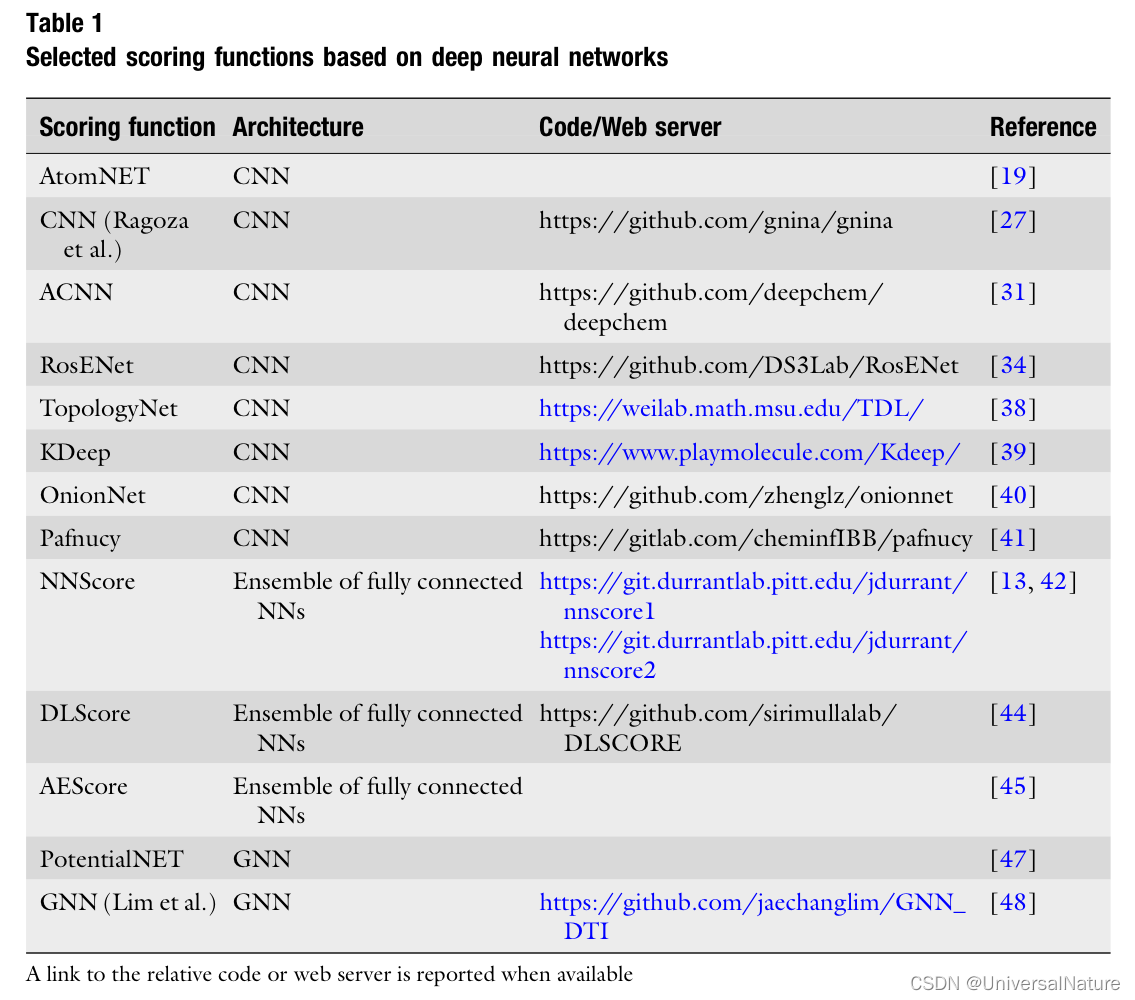
- Traditional scoring functions are grouped in force field-based, empirical, and knowledge-based. Scoring functions (Table 1) based on machine learning, nowadays, are also popular.
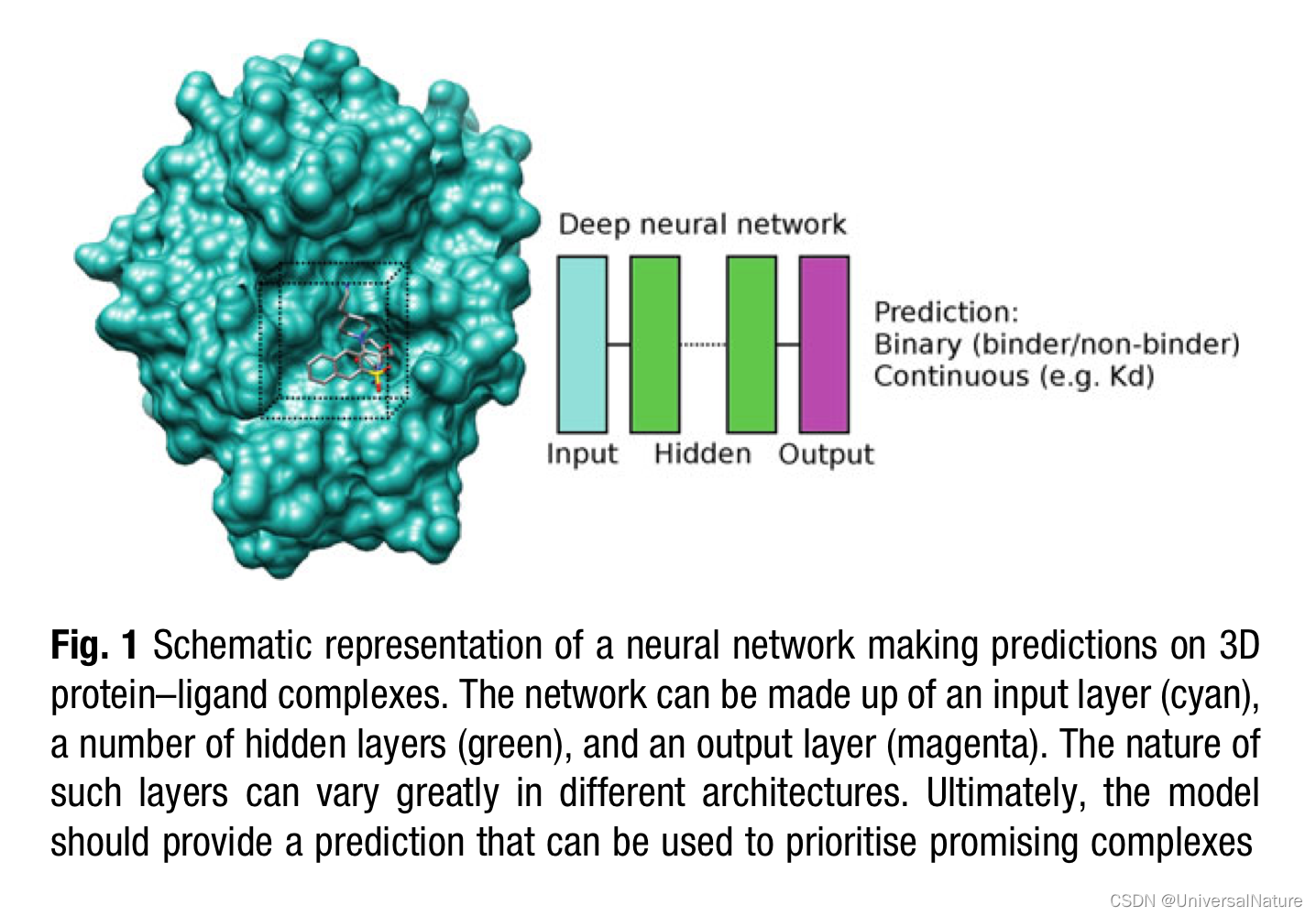
- In general, a neural network can take a representation of 3D protein–ligand complexes and be trained to predict binding affinity either as a binary classifier distinguishing binders from nonbinders or by providing a continuous value (e.g., predicted dissociation constant, Kd). A schematic view of this concept is shown in Fig. 1. Different architectures were devised for this purpose.
2.1.Convolutional Neural Networks
- CNNs applied to SBDD deal with voxels in a 3D grid which includes the ligand and the binding site residues. RGB color channels are replaced by descriptors aimed to represent the physicochemical characteristics of the protein–ligand complex.
- The first report of a CNN applied to predict binding affinity in an SBDD setup was published in 2015 by Wallach et al. Their model, named AtomNET, makes use of atom types and of different protein–ligand interaction fingerprint (PLIF) schemes to featurize input complexes.
- Ragoza et al. developed a CNN model that uses 34 distinct atom types according to the smina scheme which includes 16 receptor and 18 ligand atom types.
- the Atomic Convolutional Neural Network (ACNN) proposed by Gomes et al. directly predicts binding free energy.
- RosENet (Rosetta Energy Neural Networks) introduces MM energies as a strategy to describe protein–ligand complexes.
- Other relevant examples of scoring functions based on CNN include TopologyNet, KDeep, OnionNet, and Pafnucy.
2.2.Other Architectures
- NNScore represents an early example of scoring function based on a DNN. It was trained to distinguish binders from nonbinders by using a Kd value of 25μM as a threshold. In a later work, the improved scoring function NNScore 2.0 was described.
- DLScore uses 348 descriptors extracted with BINding ANAlyzer (BINANA).
- Recently, a scoring scheme that leverages atomic environment vectors (AEVs) named AEScore has been described.
- Graph neural networks (GNNs) have also been proposed as methods to estimate binding affinity.
- Different types of GNN have been explored. PotentialNet, for example, is based on a graph convolutional network (GCN) architecture.
- Lim et al. proposed a GNN that does not need heuristic chemical rules to deal with noncovalent interactions by devising a distance-aware graph attention (GAT) mechanism.
3.Structure-Based Virtual Screening
3.1.Score Prediction
-
Docking of a library of 170 million compounds allowed the discovery of nanomolar inhibitors of AmpC β-lactamase and of picomolar inhibitors of the dopamine D4 receptor.
-
Gentile et al. suggested a DL platform called Deep Docking which allows users to navigate ultralarge libraries by means of DNN QSAR models trained on docking scores.
3.2.Binding Mode Prediction
- Jastrze ̨bski et al. applied a DNN to predict docking poses as PLIFs. GCNs gave the most promising results.
4.Outlook
- DNN scoring functions are typically trained and validated on datasets derived from crystallographic complexes with known binding affinity and virtual screening benchmarks. Caution should be taken when using virtual screening benchmarks that include decoys as negatives.
- Indeed, some models rely extensively on information extracted from the ligand alone rather than from protein–ligand interactions.
- It has been shown that sampling wrong binding modes of active ligands as negative examples can encourage the model to take into account the ligand’s environment.
- Applications of deep learning methods related to SBDD span from the generation of relevant ligand conformations to protein structure prediction, and to binding site prediction.
















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











