







(1)从NCBI的SRA数据库查找感兴趣的的测序初始数据,这里用的是
盐胁迫下甘草地上部分药物活性成分和转录组的综合分析揭示了甘草素通过 ABA 介导的信号传导![]() https://www.x-mol.com/paper/1495711702323646464/t?adv
https://www.x-mol.com/paper/1495711702323646464/t?adv
文章里甘草(Glycyrrhiza)的SRR18163533一个样本作为例子
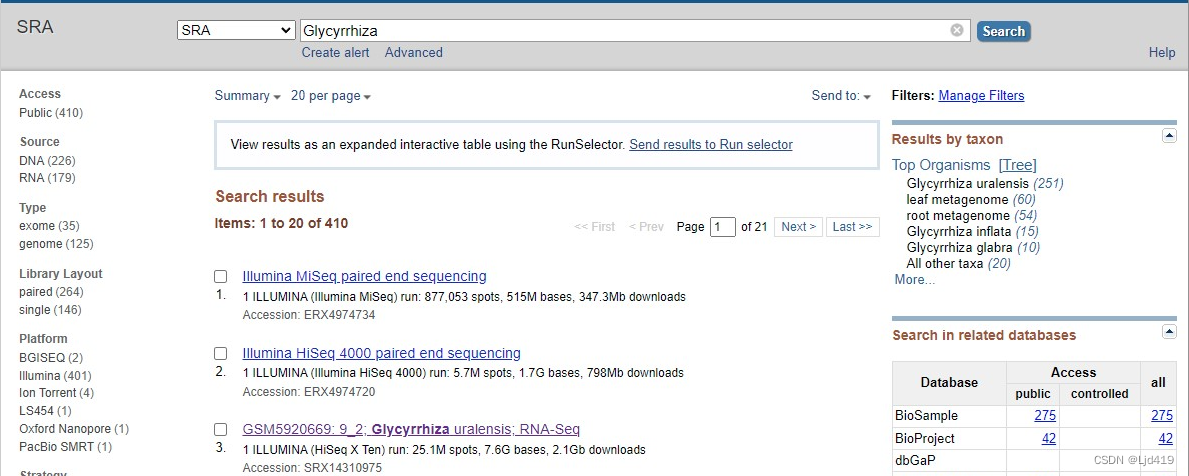
样本在第三个位置,进去的界面如下:
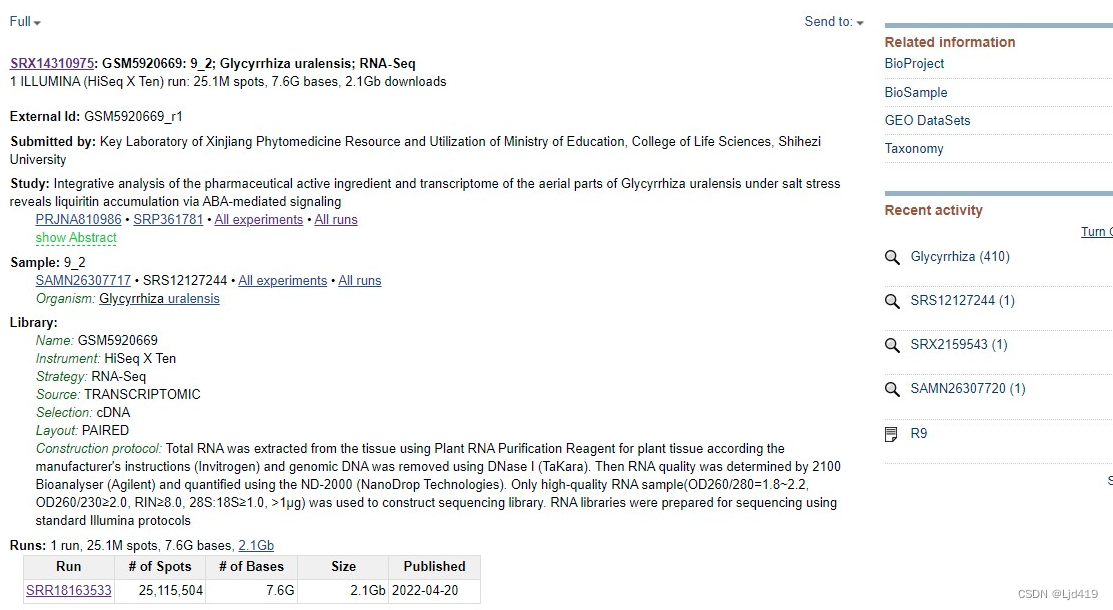
复制左下角转录组代号SRR18163533
以下处理在LINUX环境下ubuntu系统中完成
(2)SRAtoolkit软件
该软件的功能:可以从NCBI自动下载所需原始数据,且能够将sra数据转换为fastq格式
Download------Download Tools------SRA Toolkit------选择ubuntu的版本下载
可以直接在浏览器中下载,也可以复制下载所在链接在终端下载
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.0.0/sratoolkit.3.0.0-ubuntu64.tar.gz下载完成还只是安装包,需要对它进行解压,同样打开终端
tar zxvf sratoolkit.3.0.0-ubuntu64.tar.gz解压完成,但是还需要配置环境(配置环境才能让该软件能够在ubuntu中进行其功能)
echo 'export PATH=$PATH:$HOME/SRAToolkit/sratoolkit.3.0.0-ubuntu64/bin ' >> ~/.bashrc
source ~/.bashrc
vdb-config --interactive #会出现一个框架,按字母x键退出,然后就可以使用啦现在就可以用SRAToolkit软件下载原始测序数据啦!
prefetch SRR18163533 #下载.sra文件
fastq-dump SRR18163533.sra #将.sra文件转换成.fastq文件(2)质量控制和预处理
下载fastqc软件,下载步骤和上面大体一样,可以从浏览器搜索fastqc官网找到其下载地址链接下载并安装、配置环境,这里不详细赘述
fastqc -f fastq -o result SRR18163533.out.fastqfastq结果文件如下,关于fastqc所得到的这许多结果图的生物学意义自行查阅理解
(3)对读段数据进行过滤
Trimmomatic软件下载、安装等,该软件使用前需安装java,配置环境
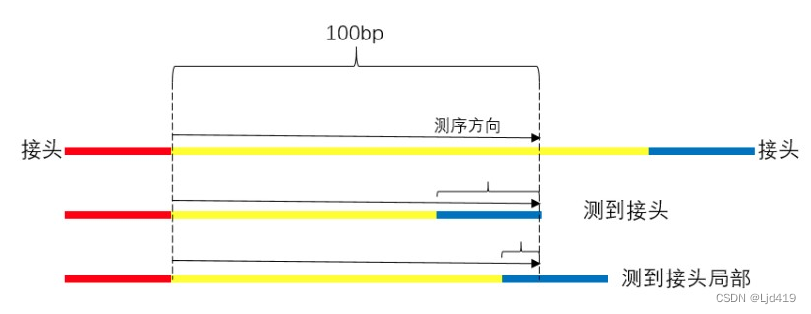
软件使用
(4)选则参考基因组并将自己的数据进行有参比对
甘草参考基因组(fasta)的下载以及其注释文件的下载(gff3)
存放在DDBJ数据库,自行下载
比对软件选择hista2,比tophat2准确且速度更快
官网下载、安装、配置环境
hisat2使用:
①构建参考索引

这里的gly-ura.fasta是我的参考基因组,代码运行后会生成许多以genome为前缀的文件
②建立各种小索引,并将读段比对到参考基因组索引
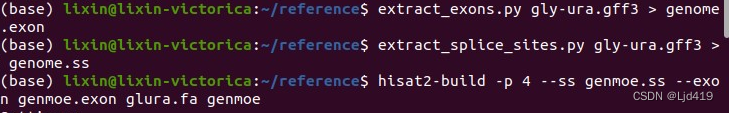
第一块代码是为了构建外显子索引空文件,第二块建立转录组索引空文件,第三块代码可以建立外显子和转录组索引文件
也可以写到一块
![]()
比对到参考基因组![]()
详细颗参考上面链接文章
(5)转录组组装
比对完成后会获得以.sam结尾的文件
需要将sam文件转换成bam文件才能进行后续处理
下载、解压安装samtools
samtools使用:
![]()
获得上面第一个文件bam结尾格式
使用samtools将.bam文件进行排序
![]()
获得上面第三个文件
使用stringtie对读段进行组装
![]()
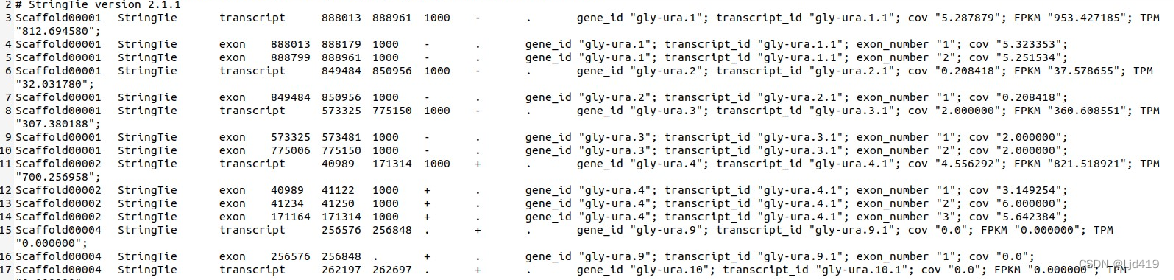
组装完成,获得转录本数据

(6)获得表达矩阵
htseq-count下载安装
htseq-count -r pos -f bam SRR18163533.bam SRR18163533.gtf > counts.txt --stranded=no















 2061
2061











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









