







标题:小麦基因组选择育种通道方法
描述:专门为小麦全基因组选择育种设计的包
编码方式:UTF-8
URL:GitHub - byzheng/BWGS: 2021 BreedWheat Genomic Selection pipeline
BugReports :https://github.com/byzheng/BWGS/issues
接下来要重点说明的是BWGS的预测:
 使用模型选择选项,仅使用基因型数据计算目标人群的GEBV预测。基本的代码如下:
使用模型选择选项,仅使用基因型数据计算目标人群的GEBV预测。基本的代码如下:
bwgs.predict( geno_train, pheno_train, geno_target, FIXED_train = "NULL", FIXED_target = "NULL", MAXNA = 0.2, MAF = 0.05, geno.reduct.method = "NULL", reduct.size = "NULL", r2 = "NULL", pval = "NULL", MAP = "NULL", geno.impute.method = "NULL", predict.method = "GBLUP" )下面是对代码中出现的各项的要求数据格式:
geno_train:
训练群体基因型矩阵(n x m):n个品系,m个标记。基因型应编码为-1,0,1,NA。缺失数据是允许的,并编码为NA。这里几个需要注意的地方是编码最好为-1,0,1,虽然编码为0,1,2本质上应该相同,训练群体应该转化为矩阵形式,这里允许有缺失数据的存在。
pheno_train:
训练表型的表型向量(n x 1)。此向量不应缺少值。否则,pheno_train和geno_train中的缺失值(NA)都将被省略。此时注意表型是向量形式,不应该有缺失值存在,要记得查看是否是向量形式。
geno_target:
目标群体基因型矩阵(z x m):具有与geno_train中相同m标记的z系。基因型应编码为-1,0,1,NA。缺失数据是允许的,并编码为NA。除了pop_reduct_method、nTimes和nFolds之外,其他参数与bwgs.cv的参数相同,因为预测只运行一次,使用整个训练群体进行模型估计,然后应用于目标群体。训练群体的基因型矩阵为 nxm,此时的目标群体的基因型矩阵的行数不用和训练群体的保证相同。
FIXED_train:
用于训练的固定效果矩阵,要与某些方法(如BGLR中包含的方法)一起使用,必须具有与geno相同的行名,并编码为(-1 0 1)
FIXED_target:
用于定位的固定效果矩阵,要与某些方法(如BGLR中包含的方法)一起使用,必须具有与geno相同的行名,并编码为(-1 0 1)
MAXNA:
geno中过滤标记列时允许的缺失值的最大比例。默认值为0.2
MAF:
基因组中筛选标记列的最小等位基因频率;默认值为0.05
geno.reduct.method:
允许对标记的子集进行采样,以加快计算时间和/或避免引入比信息标记更多的噪声。选项包括:
RMR:标记子集的随机抽样(不替换)。与参数“reduct.marker.size”一起使用
中间这些参数都不是非必须,一般用不到,下边我们直接来看一下本文用到的方法。
predict.method:
基因组育种价值预测方法的选择。可用的选项有:
GBLUP:使用基于标记的关系矩阵执行G-BLUP,通过BGLR R-library实现。相当于标记效应的岭回归(RRBLUP)。
EGBLUP:执行EG-BLUP,即BLUP使用“平方”关系矩阵来模拟上位2x2相互作用,如Jiang&Reif(2015)所述,使用BGLR库
RR:岭回归,使用包glmnet。理论上严格等同于gblup。
LASSO:最小绝对收缩和选择算子是另一种惩罚回归方法,它产生比RR更多的收缩估计。由glmnet库运行。
EN:弹性网(Zou和Hastie,2005),它是RR和套索的加权组合,使用glmnet库
几种贝叶斯方法,使用BGLR库:
BRR:贝叶斯岭回归:与rr-blup相同,但贝叶斯分辨率。以高斯分布诱导所有标记效应向零的均匀收缩(de los Campos等人,2013年)
BL:贝叶斯LASSO:在标记方差先验上使用指数先验,导致标记效应的双指数分布(Park&Casella 2008)
BA:贝叶斯A使用标记效应的标度先验分布。
BB:Bayes B,使用点质量为零的混合分布和具有标度t分布的非零标记效应板(Habier等人,2011年)。
BC:贝叶斯C与贝叶斯B相同,具有高斯分布的板。
有关这些方法的更详细描述,请参见Perez&de los Campos 2014(http://genomics.cimmyt.org/BGLR-extdoc.pdf)。
三种半参数方法:
RKHS: reproductive kernel Hilbert space and multiple kernel MRKHS, using BGLR (Gianola and van Kaam 2008).基于遗传距离和核函数来调节标记效应的分布。这种方法被认为是检测非加性效应的有效方法。
RF:随机森林回归,使用随机森林库(布雷曼,2001年,布雷曼和卡特勒2013年)。该方法使用基于bootstrapping数据的树节点上的回归模型。应该能够捕捉到标记之间的相互作用
SVM:支持向量机,由e1071库运行。有关详细信息,请参见Chang,Chih-Chung和Lin,Chih-Jen:LIBSVM:支持向量机库http://www.csie.ntu.edu.tw/~cjlin/libsvm
BRNN:前馈神经网络的贝叶斯正则化,带有R包BRNN(Gianola等人,2011年)。To keep computing time in reasonable limits, the parameters for the brnn function are neurons=2 and epochs = 20.
Value:
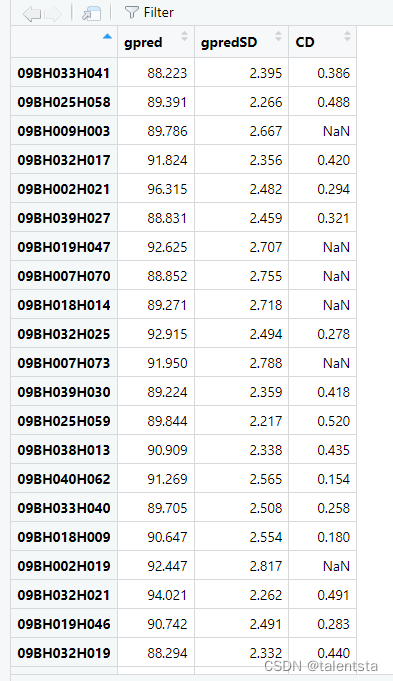
对象bwgs.predict返回维数为nx3的矩阵。列包括:
预测BV:验证集的GEBVs的nx1向量(geno_valid的行)
gpredSD:估计GEBV的标准偏差
CD:每个GEBV的决定系数,估计为sqrt((1-stdev(GEBVi))^2/2g)
请注意,gpredSD和CD仅适用于使用BGLR库的方法,即GBLUP、EGBLUP、BA、BB、BC、BL、RKHS和MKRKHS。这两列包含方法RF、RR、LASSO、EN和SVM的NA。
data(inra) # Prediction using GBLUP method
predict_gblup <- bwgs.predict(geno_train = TRAIN47K, pheno_train = YieldBLUE, geno_target = TARGET47K, MAXNA = 0.2, MAF = 0.05, geno.reduct.method = "NULL", reduct.size = "NULL", r2 = "NULL", pval = "NULL", MAP = "NULL", geno.impute.method = "MNI", predict.method = "GBLUP")
这是代入数据集的使用方法,大家可以通过这里查看数据集的格式,从而改变你自己的数据集的格式用于分析。

Description:
inra数据包含一组geno47K(760 x 47839)、pheno(760 x 1)和MAP47K(47839 x 3)。表型表型包含多年/地点试验中产量性状(YLD)的调整基因型平均值。
inra数据包含一组geno47K(760 x 47839)、pheno(760 x 1)和MAP47K(47839 x 3)。表型表型包含多年/地点试验中产量性状(YLD)的调整基因型平均值。此时可以看到过程中分别用到的数据的格式。
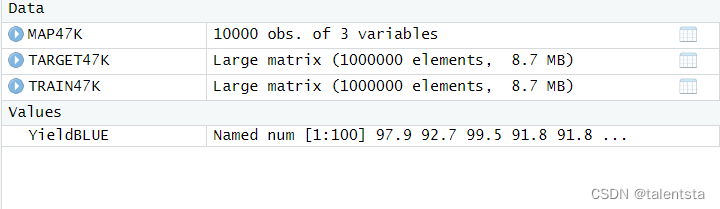
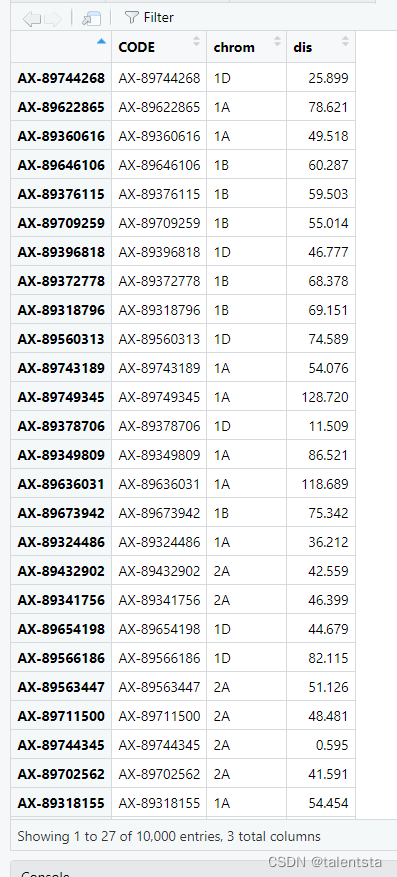
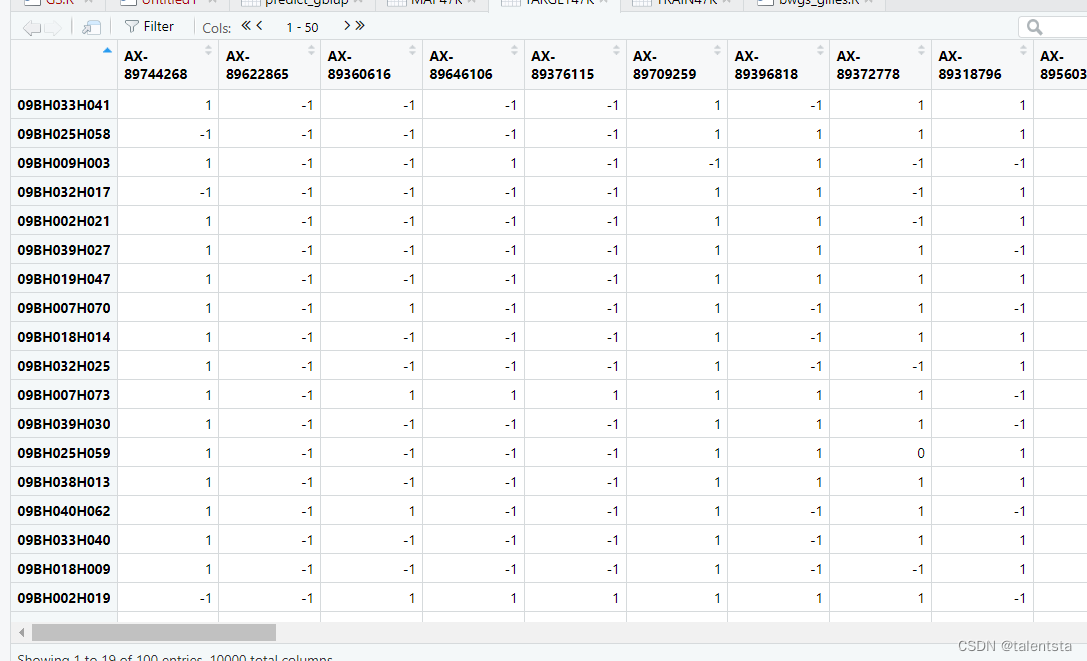
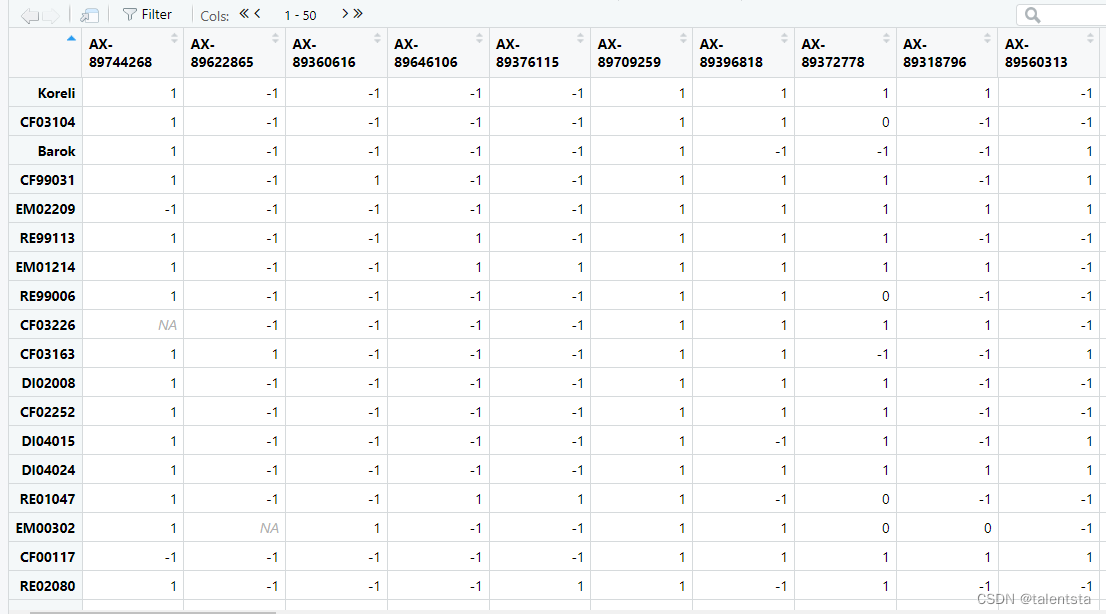 以上是按照目录顺序依次查看三个表格的结果,下边是产生的预测值
以上是按照目录顺序依次查看三个表格的结果,下边是产生的预测值
Format:
类的对象数据帧10000行3列。
matrix类的对象(继承自array),有100行10000列。
matrix类的对象(继承自array),有100行10000列。
长度为100的类numeric对象。















 1589
1589











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









