







点击蓝字 · 关注我们

论文ID
原题:Machine learning and metagenomics reveal shared antimicrobial resistance profiles across multiple chicken farms and abattoirs in China
译名:机器学习和宏基因组学揭示了中国多个养鸡场和屠宰场共同的抗菌素耐药特征
通讯作者:Zixin Peng*, Fengqin Li* & Tania Dottorini*
研究机构:
1、School of Veterinary Medicine and Science, University of Nottingham, Sutton Bonington, UK
2、NHC Key Laboratory of Food Safety Risk Assessment, China National Center for Food Safety Risk Assessment, Beijing, People’s Republic of China
期刊:Nature Food
发表时间: 2023

研究背景
01
中国禽类生产中的抗菌药物使用量比国际平均水平高出五倍。即使在低水平下使用抗生素也会改变和扩展家畜的肠道耐药基因组,微生物群落也会形成抗菌素耐药(antimicrobial resistance, AMR)表型。饮食、温度和压力等外部因素可能导致新的细菌种群定居或不同物种之间的抗药性转移。温度、湿度、细菌物种丰度以及抗生素耐药基因的存在都可以影响肉鸡的细菌感染。对于中国和低收入和中等收入国家,环境条件与抗菌药物抗性之间的关联尤为重要。相较于高收入国家,这些国家在工业化规模的农业生产中维持稳定的环境条件可能具有挑战性。
虽然非卫生领域的抗菌素耐药监测并不普遍,但是它对于了解食品生产系统如何促进抗生素耐药菌和抗生素抗性基因的选择和传播至关重要。机器学习和大数据挖掘为提高家禽精细化养殖提供了工具。基于培养的方法涉及对单个病原体的全基因组测序、抗生素敏感性测试和机器学习技术,对大肠杆菌分离株和其他细菌的基因组特征与抗生素抗性之间的联系进行了有效预测。然而,仅关注单个病原体的全基因组测序的监测方法可能无法捕捉到家畜生产中的微生物群落和耐药基因组的多样性,可能会遗漏一些抗生素抗性基因数据。在最近的概念验证研究中,我们观察到鸡粪便耐药基因组中存在的一些耐药基因与同一样品中培养出的大肠杆菌株的耐药/敏感性特征有关。
在本研究中,作者开发了一种针对中国养殖业的基于宏基因组的监测方法。在中国和中低收入国家中,进行抗菌药物抗性监测尤为具有挑战性,因此作者考虑到了常见的实验室资源匮乏问题。以大肠杆菌作为指示物种,以更广泛的鸡肠道微生物群落为背景来评估抗菌素耐药情况。为了考虑更广泛的背景,作者研究了养殖场周围相连的环境、仓库温湿度以及抗菌药物的使用方案对微生物组的影响。
材料和方法
02
在本研究中,作者选择了中国三个不同省份(山东、河南和辽宁)的十个大型商业禽类养殖场(下文简称为SDx、HNx和LNx),覆盖面积为472,500平方千米。每个养殖场都供应给四个地区性的屠宰场之一(河南有两个,辽宁有一个,山东有一个)。每个养殖场都有多个存栏,每个存栏容纳12,000到32,800只禽鸟,每个繁育周期的总产能为110,730到380,000只禽鸟。肉鸡的繁育基于自繁自养的模式,禽鸟在养殖场繁育后以同龄批次转移到饲养舍内。在选定的十个养殖场中,有四个(辽宁的三个,山东的一个)使用网式养殖系统,而其他六个使用笼式养殖系统。在样品收集期间,两种饲养系统之间的每个饲养舍内禽鸟数量差异不显著(t检验,P = 0.07)。
除了在山东省的一个养殖场进行两个繁育周期的采样以进行试点研究,对采集活动和数据分析规程进行微调外,其它养殖场均在一个繁育周期内对同一批次的禽鸟进行采样。生物样品在每个繁育周期的三个时间点进行采集:t1(第3周),t2(第6周)和t3(第6周后1-5天)。生物样本包括在活禽生命中期(t1)和生命末期(t2)从畜舍地面采集的活禽排泄物中的粪便和羽毛,以及在相同时间点采集的畜舍地面样本。在屠宰场,屠宰当天(t3)从宰杀动物的胴体、肉类加工表面(称为加工线)和废水中采集样本。从围绕养殖场的外部区域采集土壤样本(t1和t2)。
大多数养殖场(HN1、HN2、HN3、SD2、SD3和SD4)使用自动化传感器和数据记录仪,以5分钟的时间间隔收集环境传感器数据(温度和湿度)。三个养殖场(SD1、LN2和LN3)手动测量,每天或每6小时测量一次。
生物样品经DNA提取和文库构建并测序后,原始序列数据进行组装和分箱等生物信息学分析。
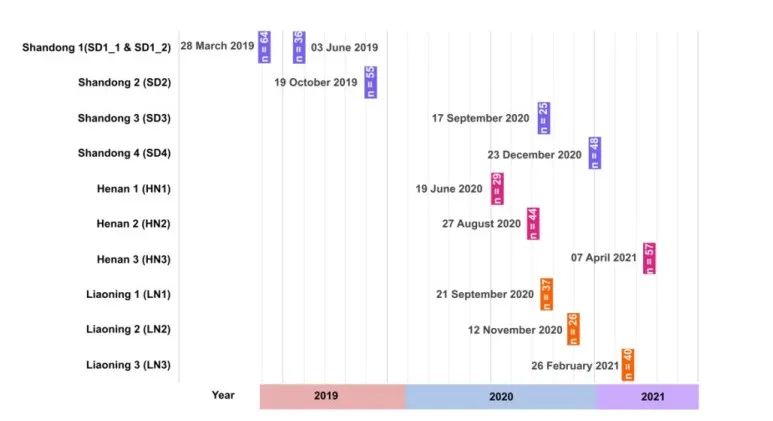
图1. 生物样品和环境样品总结。A样品采集的省份。B 生物样品采集的来源和时间周期节点。C 每个养殖场样品收集的时间。
结果和讨论
03
3.1禽类和环境共享临床相关的可移动抗生素抗性基因
从十个大规模商业家禽养殖场收集了生物样品进行生物信息学分析。微生物群落和抗生素抗性基因在养殖场和屠宰场之间,以及不同养殖场之间存在差异。由于基因迁移可能影响不同来源抗生素抗性基因的存在,并且可移动遗传元件(MGEs)在开发有效监测系统中具有潜在重要性,作者检索了与MGEs相距5千碱基对(kb)范围内的抗生素抗性基因(ARGs),并将这些MGE-ARG组合视为潜在的可移动抗生素抗性基因。总共发现了661个不同的MGE-ARG组合(潜在可移动抗生素抗性基因),其中包含了195个独特的抗生素抗性基因。其中75个抗生素抗性基因(38%)仅在一个MGE-ARG组合中发现,而其余的120个(62%)在多个组合中发现(2到22个;图2a)。超过一半(56%)的潜在可移动抗生素抗性基因在多个来源中存在(图2b),其中三个MGE-ARG组合(IS1216-poxtA、IS15-APH(3')-Ia和ISCfr1-AAC(3)-IId)在除羽毛以外的所有来源中均存在。鸡粪中潜在可移动抗生素抗性基因数量最多,但差异也最大(图2c)。羽毛和谷仓地面也携带了许多潜在可移动抗生素抗性基因,平均数量在统计上与粪便相当(Dunn's test调整后P > 0.05)。户外土壤、屠宰胴体、加工线和废水样品通常具有较低数量的潜在可移动抗生素抗性基因,其数量与粪便和羽毛有显着差异(Dunn's test调整后P < 0.01),但与彼此间没有差异。在所有十个农场中,在同一农场的禽鸟和环境来源中共发现了145个不同的MGE-ARG组合,其中一些出现在多个农场。其中46个包含临床相关的抗生素抗性基因(图2d)。值得注意的是,我们在鸡粪便、羽毛和环境谷仓地面样品中发现了blaNDM-5基因。该基因通常存在于IncX3质粒上,在人类、动物、食品和环境中广泛传播,尽管我们并未在短读宏基因组测序(MGS)数据中证实质粒的存在。另一个重要的临床相关基因qnrS1在鸡粪便、羽毛、环境谷仓地面和废水样品中发现。这种质粒介导的喹诺酮类耐药基因已知存在于鸡肉供应链中,并能够转移到不同的细菌中。
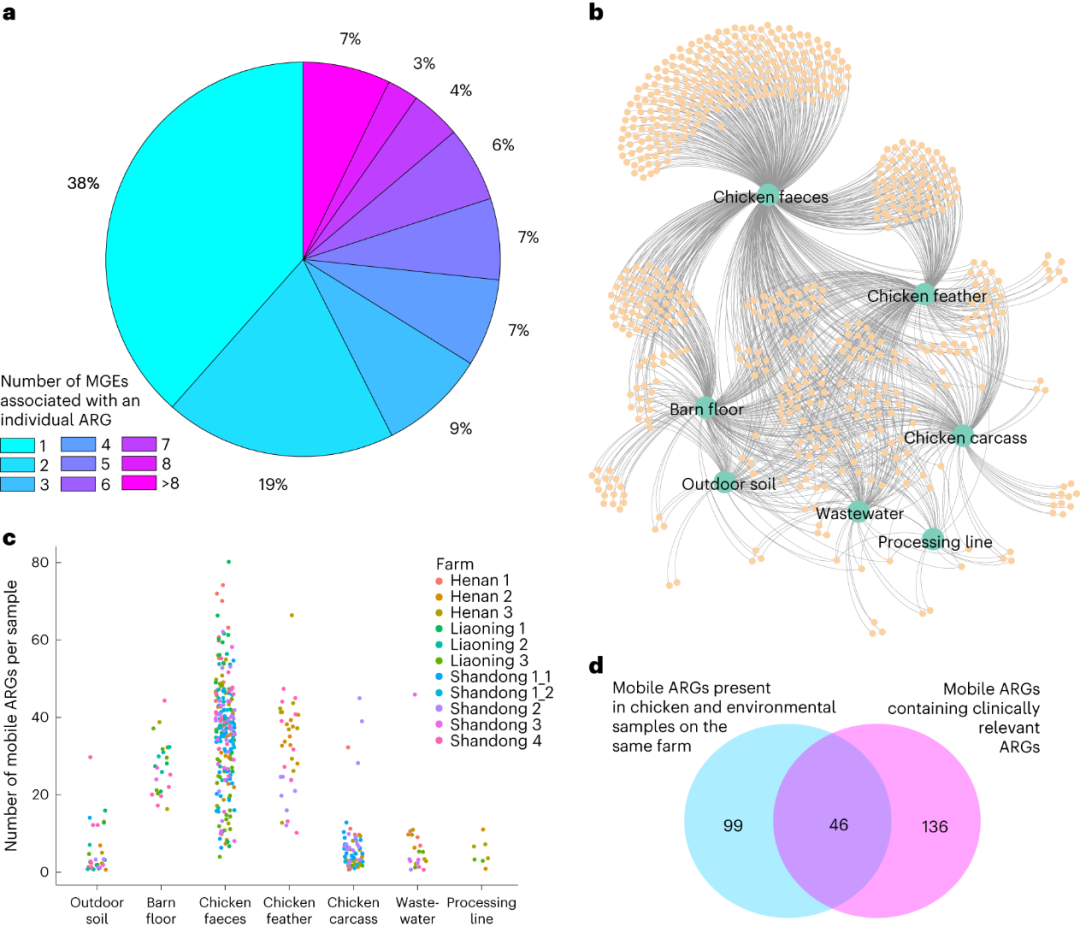
图2 潜在可移动ARG的分析。a,饼图显示与一个或多个 MGE 相关的 ARG(在已发现的 195 个中)的比例。b,无向网络图,显示与不同样品源(绿色大圆圈)相关的潜在移动ARG(橙色小圆圈)。图中的边将潜在的可移动 ARG 连接到它们所在的源。c,每个来源的每个样品的潜在可移动ARG数量。每个圆圈代表一个样品,圆圈按农场着色。d,维恩图显示,在同一养殖厂的鸡和环境样品中发现了145个(共661个)潜在可移动ARG,182个潜在可移动ARG包含临床相关的ARG。从同一养殖场获得的鸡和环境来源中发现了46 个潜在的临床相关移动ARG。
3.2大肠杆菌抗菌素耐药与其栖息的肠道微生物组相关
作者进一步调查了鸡肠道中发现的细菌种类,抗性组和从相同样品中获取的大肠杆菌分离株的抗菌素耐药谱之间是否存在相关性。作者从170个鸡粪便样品中培养了大肠杆菌分离株,并对26种抗生素的抗菌素耐药性进行了表征。对每种抗生素耐药的菌株的比例在1%到98%之间变化。所有分离株至少对一种抗生素具有耐药性,其中169株对至少三种抗生素具有耐药性。
为了研究大肠杆菌中抗生素耐药性与肠道微生物组之间的相关性,作者开发了一种基于机器学习的定制数据挖掘方法(图3)。该方法包括构建一种机器学习驱动的“预测函数”,其输入是来自肠道微生物群落(微生物物种的相对丰度)和肠道耐药组(ARG计数)的信息聚合,其输出是大肠杆菌对特定抗生素的抗性(是或否),该输出基于抗微生物药物敏感性测试(antimicrobial susceptibility testing, AST)。通过使用实验数据进行训练(监督学习)并交换不同的基础机器学习技术,直到达到最佳的预测性能为止。从机器学习模型中提取了一组最具信息量的特征,也称为“预测因子”。然后通过与温度和湿度的相关性分析对该集合进行了改进。
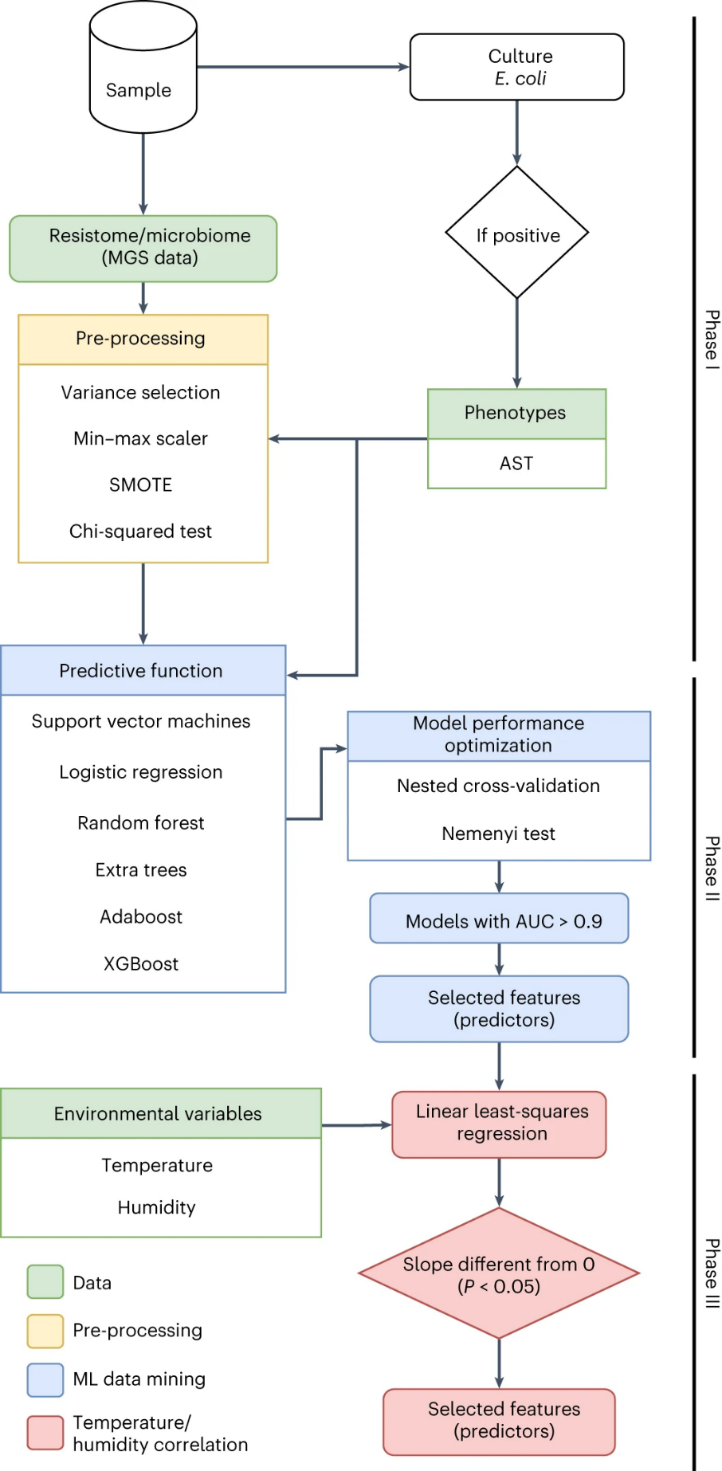
图3 通过数据挖掘流程查找肠道微生物组、大肠杆菌中的抗生素耐药性、温度和湿度之间的相关性。基于ML的定制数据挖掘方法的完整数据分析工作流程,输入数据以绿色显示。第一阶段涉及宏基因组数据预处理(黄色)。第二阶段涉及训练和测试ML驱动的预测功能,以分离与表型抗性(蓝色)相关的宏基因组特征(即样品中存在的微生物物种的ARG计数和相对丰度)。第三阶段涉及拟合回归模型(在下一节中讨论),以分离与温度和湿度变化(红色)更好地相关的宏基因组特征。AUC,曲线下面积。
在26种抗生素中,只有17种有足够的数据(耐药性和敏感性病例)进行适当的ML训练。数据挖掘显示,鸡肠道核心抗性组(通过鸡粪便宏基因组数据所有检测到的抗生素抗性基因)和微生物种类(通过鸡粪便宏基因组数据检测到的所有细菌物种)对大肠杆菌耐药性具有较强的预测能力。共计包括419个特征(186种微生物物种和233个抗生素抗性基因),作为大肠杆菌对10种抗生素(图4b、c)的耐受性/敏感性的强力预测因子,AUC值超过0.90。前10种抗生素模型中的233个耐药基因分别属于β-内酰胺类药物(24%的基因)、氨基糖苷类药物(18%)和大环内酯类、林可胺类和链霉素类B(MLSB;18%),其他抗生素类药物的比例不到10%。在这233个ARGs中,根据耐药基因短读序列数深度与物种丰度的相关性,发现46个ARGs源自大肠杆菌。另外16个耐药基因仅存在于被识别为其他细菌物种的连续序列中(即它们并非源自大肠杆菌)。为了进一步探索肠道核心特征与抗生素抗性之间的关系,将这419个特征和10种抗生素抗性可视化为一个图的节点,边仅连接预测因子和被预测的抗药性(图4c)。该分析突显了一个包含66个ARGs(其中15个在临床上具有重要意义,包括blaNDM-5、blaCTX-M-15、dfrA15和dfra5)的核心,作为超过3种抗生素抗性的预测因子。三个ARGs(aphA6、vat(A)和vgb(A))被发现是8种抗生素抗性的预测因子。同样的分析还揭示了肠道中的28种微生物物种作为5种抗生素抗性(阿奇霉素、氯霉素、头孢噻肟、卡那霉素和萘啶酸)的预测因子。这28种物种包括细菌属弧菌、不动杆菌和弯曲杆菌,以及其他共生细菌。

图4 在大肠杆菌中,通过肠道微生物物种、抗性组和抗生素耐药性之间的相关性进行ML性能和特征选择。a,机器学习驱动的大肠杆菌对特定抗生素耐药性的预测功能的性能。b,宏基因组特征(ARGs和微生物物种)计数是大肠杆菌对每种抗生素耐药/敏感概况的最强预测因子。c,无向图显示每个抗生素模型的最强预测因子(鸡肠道中的宏基因组特征)。图形的边将ARG或细菌种类节点(预测变量)连接到发现它们具有预测性的抗生素模型。ARG 和抗生素模型节点均根据已知与抗生素/ARG 相关的抗生素类别进行颜色编码。
对于排名前十的抗生素模型,作者开发了定制的回归模型,使用单个肠道特征作为自变量(每个变量一个模型),温度或湿度作为因变量,以确定模型拟合是否会突出相关性。在原有的419个特征中,130个ARGs和48个微生物物种与湿度相关,39个ARGs和20个微生物物种与温度相关。作者测试了一些与温度或湿度相关的ARGs可能属于也与温度和湿度相关的微生物物种的可能性。分析突出显示了与湿度相关的两个不同的子图(图 5a)和一个与温度相关的子图(图 5b). 值得注意的是,其中一个与湿度相关的子图包含肺炎克雷伯菌和四个相关的 ARGs(kpnE、kpnF、kpnG和acrA)。在两个分析中都发现了含有粪芽孢杆菌和ARGs vga(C)和blaOXA-58的子图(即它与温度和湿度都相关)。
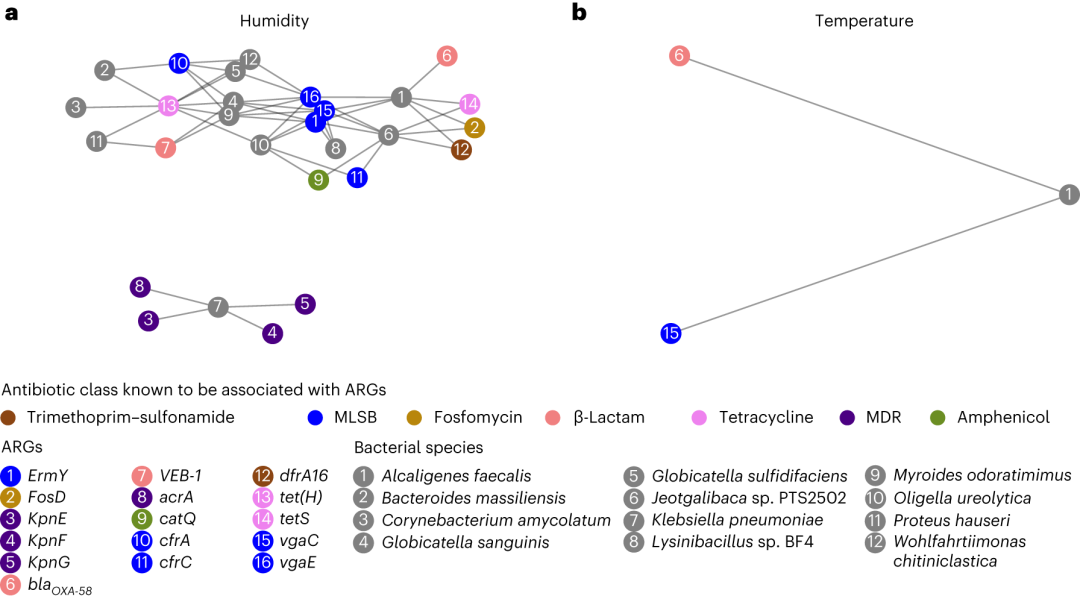
图5 肠道特征被确定为大肠杆菌耐药性的预测因子。a,b, 微生物种类和ARGs与湿度(a)和温度(b)相关。微生物种类和ARGs与湿度或温度相关,且彼此相关,表明ARGs可能存在于物种中。如果线性回归线的斜率显著不同于零,则认为特征相关(使用双侧t检验的P < 0.05)。节点表示ARGs或微生物物种;边将物种与物种中可能存在的ARGs连接起来。ARG 节点根据已知与 ARGs 相关的抗生素类别进行颜色编码;微生物物种节点以灰色显示。
然后作者调查了肠道中作为大肠杆菌耐药性预测因子且与温度和湿度相关的耐药基因特征是否与可移动遗传元件紧密相连。在靠近可移动遗传元件附近位置找到了10个耐药基因(MLSB:optrA、mph(F)和erm(X);β-内酰胺:blaNDM-1和blaOXA-58;氨苄菲:catA8和catB2;氨基糖苷类:aadA1;氟喹诺酮:qnrS2和qnrA1)。这10个耐药基因中的3个只与1个可移动遗传元件相关联(catB2与ISPa25,mph(F)与IS15和qnrA1与IS15),而其他7个与2-9个不同的可移动遗传元件相关联。对于所有的可移动遗传元件-耐药基因对,进行了跨农场或来源的保守结构调查。例如,临床重要的blaNDM-1在四个样品中与IS15紧密相连(LN1的三个鸡粪便样品和辽宁3号(LN3)的一个畜舍地面样品)。在来自LN1、LN3、山东2号(SD2)和山东4号(SD4)的鸡粪便和羽毛样品中发现,blaNDM-1靠近可移动遗传元件 ISAba125,并且紧邻另一个耐药基因ble,在亚洲地区大肠杆菌科物种中质粒携带blaNDM-1是已知的关联。尽管在多个农场(LN1、LN3、SD2和SD4)中发现了相同的blaNDM-1-ISAba125模式,但没有证据表明其在农场之间传播(图6)。相反,对连续序列进行了进化分析(使用分子钟模型预测系统发生树每个分支上分子进化速率),提示单个农场内分离株的分支较近(大多数分支的最近公共祖先(MCRA)不超过2年),以及不同农场之间最近公共祖先则更早(大于20年),表明这种潜在的可移动ARGs在中国各地的家畜中广泛传播的可能性。
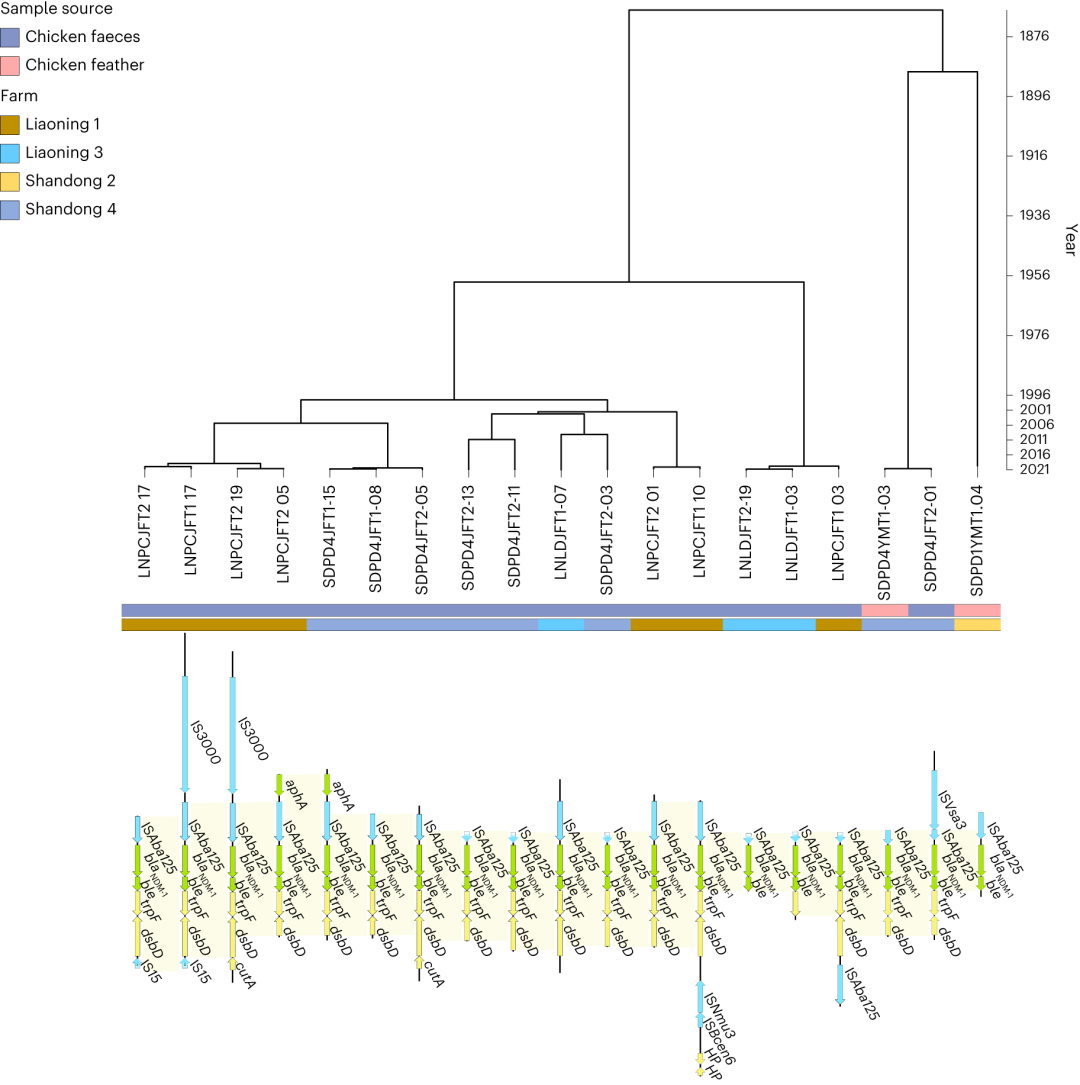
图6 潜在可移动ARG模式ISAba125-NDM-1的基因结构和进化分析。贝叶斯进化系统发育树重建含有临床重要ARG blaNDM-1和MGE ISAba125的contigs的系统发育。
总结
04
传统的AMR监测方法无法准确评估养殖场内和养殖场之间的细菌和耐药组多样性。大肠杆菌作为AMR的参考指示指标已经在之前被应用。在本研究中,作者使用大规模宏基因组采样和大肠杆菌分离,结合统计和机器学习方法绘制AMR趋势和模式的复杂相关性,发现38种临床相关的ARGs与对多种抗生素的耐药性相关,其中一些抗生素与这些ARGs之前没有已知的相关性。研究先一组肠道细菌与大肠杆菌对5种不同抗生素的耐药性密切相关,包括弓形菌属、不动杆菌和鞘杆菌,这表明专注于大肠杆菌不如监测更多数量的病原体有效。
宏基因组测序拓宽了我们对耐药性驱动因素的了解并改善AMR监测。耐药性产生于抗生素耐药微生物、微生物群落、地理生态位和环境、进化力量、气候和人类实践之间的复杂相互作用。需要进一步推动能够监测AMR动态的监测解决方案的创新和开发。
*本文如有不足,请批评指正
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









