






点击蓝字 关注我们
解译恢复草地土壤微生物生活史策略及其影响因素


iMeta主页:http://www.imeta.science
研究论文
● 期刊:iMeta(IF 23.8)
● 文章被引(Dimensions截至2025年2月5日): 109
● 原文链接DOI: https://doi.org/10.1002/imt2.66
● 2022年12月5日,中科院地理环境研究所王云强团队等在iMeta在线发表了题为“Deciphering factors driving soil microbial life-history strategies in restored grasslands”的文章。
● 本研究构建了一个框架,以突出草地恢复过程中植物和土壤特性在驱动微生物生活史特征方面的重要性,表明微生物生活史特征支持rRNA操纵子拷贝数能够反映土壤微生物资源有效性这一观点。
● 第一作者:杨阳
● 通讯作者:王云强 (wangyq@ieecas.cn) 、安韶山 (shan@ms.iswc.ac.cn)
● 合作作者:窦艳星、王宝荣、薛志靖、Scott X. Chang
● 主要单位:西北农林科技大学、陕西师范大学、加拿大阿尔伯塔大学
亮 点
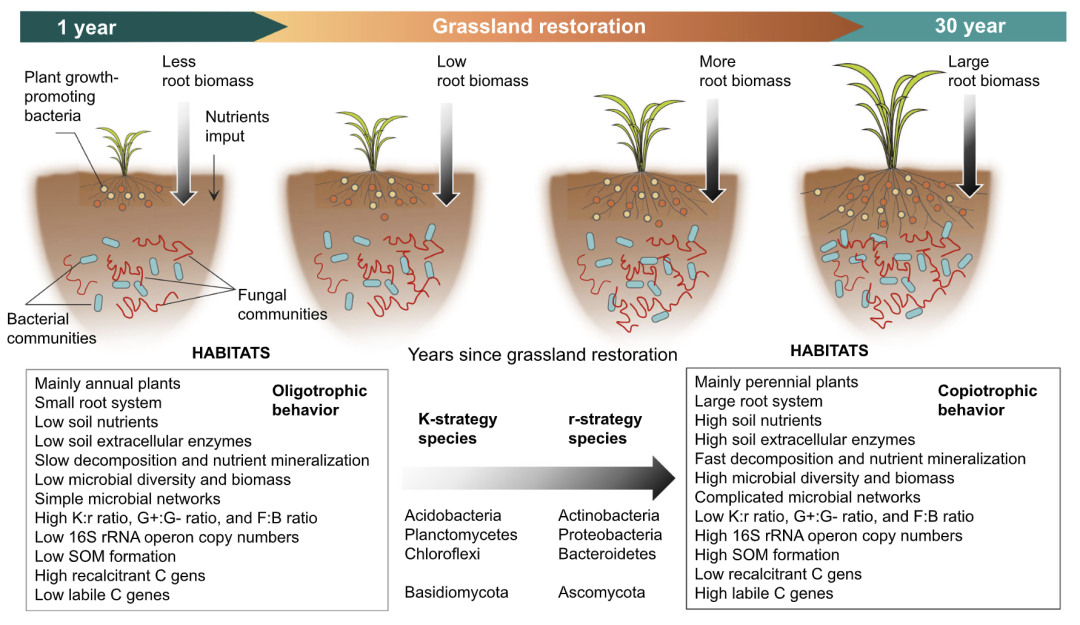
● 草地恢复增加了r-对策的土壤微生物相对丰度,减少了K-对策的土壤微生物相对丰度
● 草地恢复导致土壤微生物群落由寡营养型向共营养型的转变
● 土壤性质与土壤微生物生活史性状呈正相关性
● 植物属性与土壤微生物生活史性状呈负相关性
摘 要
在宏观生态学中,虽然r-和K-策略的概念已被广泛应用,但是运用多种测序方法探明温带草原微生物生活史策略的研究较少。通常用总磷脂脂肪酸分析、高通量宏基因组测序和GeoChip技术来检验草地不同恢复年限(恢复1、5、10、15、25和30年)微生物生活史特征的变化。草地恢复增加了放线菌、变形菌和拟杆菌的相对丰度,但减少了酸杆菌、浮游菌和绿弯菌的相对丰度。磷脂脂肪酸分析表明,草地恢复降低了真菌:细菌比、革兰氏阳性菌:革兰氏阴性菌比。结合宏基因组数据,我们发现草地恢复将微生物从寡营养型(K-)转变为富营养型(r-),与微生物群落rRNA操纵子拷贝数的增加一致。结构方程模型表明,微生物生活史特征与土壤特性正相关(P<0.05),而与植物特性负相关(P<0.01)。我们构建了一个框架,以突出草地恢复过程中植物和土壤特性在驱动微生物生活史特征方面的重要性。最后结合宏基因组和其他微生物数据,我们的研究结果表明微生物生活史特征支持rRNA操纵子拷贝数能够反映土壤微生物资源有效性这一观点。
视频解读
Bilibili:https://www.bilibili.com/video/BV1244y1o7L4/
Youtube:https://youtu.be/LPT0ZVUgZZM
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
土壤生物中占据绝大多数的是微生物,其在陆地生态系统中非常复杂。基于现有的生态学理论,微生物生态学家提出了基于性状的土壤微生物分类方法。富营养-寡营养分类法被认为类似于植物、动物的r-和K-选择理论。该分类法主要基于微生物底物偏好、营养策略和生长速率,已广泛应用于各种环境中。以往的研究将竞争-胁迫耐受-传播定植(Co-S-R)生活史策略应用于微生物系统,特别是在人为环境变化的背景下。由于Co-S-R方法允许使用混合生活史策略(如CoS、SR等)对微生物进行分类,它已被广泛应用于微生物生态学研究。最近的研究在Co-S-R分类框架的基础上提出了一个修订的生活史策略,并指出三种主要的微生物生活史策略:高生长量(Y)、资源获取(A)和胁迫耐受(S),或Y-A-S,两个主要的环境变量:资源有效性和非生物胁迫。例如,Y-策略微生物有效地将单体底物(如葡萄糖)转化为微生物生物量,然后再到微生物残体。相反,A-策略微生物在资源有效性较低的情况下占优势,此时,微生物面临着以牺牲生长为代价来获取资源。
土壤微生物能够适应营养丰富和贫乏的环境,从而使得微生物活性与碳之间的关系变得复杂。土壤中微生物的生长和功能受到底物数量和质量的严重影响。微生物群落使用多种生活策略来响应,进而影响土壤碳的动态。研究表明,微生物群落组成与其有效性或底物利用策略有关。根据微生物碳矿化潜力和生长速率,可以分为两个生态功能类别,即r-和K-策略。r-策略物种(富营养或机会物种)生长速度快,底物亲和力低,对有效碳和养分输入反应迅速,通常在富含不稳定碳的环境中生存。相反,K-策略物种(寡营养或平衡物种)生长缓慢,具有较高的底物亲和力,能有效地利用难分解的碳。为探明微生物生长、碳循环和资源获取之间的关系,富营养微生物(r-策略)和寡营养微生物(K-策略)可以用来预测微生物活性及其与环境的相互作用,因为这些过程在很大程度上控制了碳循环。
随着基因组测序技术的进步,微生物生活史特征已广泛应用于各种生态系统。基于宏基因组测序,rRNA操纵子拷贝数可反应微生物群落特征,识别出r-或K-策略微生物。通常,拥有较多rRNA操纵子拷贝数的微生物被视为r-策略,因为rRNA操纵子的数量与最大生长率、生长速率转变力、以及少部分的高亲和转运蛋白相关。富营养菌的快速生长需要细胞核糖体的大幅增加,主要是通过基因组中rRNA操纵子拷贝数的增加来实现的。相反,拥有一个或几个rRNA操纵子拷贝数的微生物被视为K-策略,从有限的资源中获取营养。寡营养菌的高效生长使得消耗单位资源繁殖更多的后代,并导致基因丢失和rRNA操纵子拷贝数减少。此外,快速生长的富营养菌也会增强其核糖体基因中同义密码子的使用,因为它们经历了翻译选择,导致更大的密码子使用偏性。因此,更大的密码子使用偏性通常与更高的最大生长率有关,这代表了富营养菌对资源脉冲的响应能力。
由人类活动引起的土地退化分布广泛,是环境保护的一大挑战。黄土高原正面临严重的土地退化问题,并且由于地形破碎和人为干扰,容易受到侵蚀。为恢复植被,减少水土流失,保护生态环境,自1999年开始实施“退耕还林”政策。草地恢复在西北地区应用广泛。这些举措改变了土壤微生物群落的结构和组成。由于研究微生物生活史策略的方法不同,土壤微生物在植被恢复过程中呈现出不同的生活史策略。先前的荟萃分析和实验研究表明,在废弃农田的自然演替过程中,真菌与细菌比显著增加,因为细菌通常被视为r-策略菌,真菌被视为K-策略菌。例如,Zechmeister-Boltenstern发现,在植被演替过程中,细菌群落从r-策略转变为K-策略,真菌丰度、真菌:细菌比随着演替而增加。同样,在辽东栎次生演替中,土壤微生物群落在门和属两个层次上都倾向于从r-策略转变为K-策略。尽管Zhang等人指出,在30年的植被演替中,细菌群落从以酸杆菌为主(生长缓慢的寡营养菌,K-策略菌)转变为以变形杆菌为主(生长迅速的富营养菌,r-策略菌)。因此,正如测序方法所揭示的,不同生态系统类型之间的土壤微生物生活史策略存在很大差异。然而,在温带草原,缺乏用多种测序方法研究微生物生活史策略。植物和土壤特性对微生物生活史特征的影响的研究,对于揭示它们如何调节陆地生态系统中的生物地球化学循环至关重要。
通过概念模型来表明草地恢复过程中植物和土壤特性对微生物生活史策略的影响(图1)。土壤有机碳(SOC)是由微生物转化植物凋落物而形成,细菌和真菌是分解SOC的主要贡献者,取决于养分有效性。草地恢复会直接改变凋落物、根系、根系分泌物和土壤性质,进而改变当地的环境条件。随着草地恢复年限的增加,植物根系会产生几种信号物质和化合物来保护自己。根系分泌物中的大多数化合物可以促进微生物生物量和胞外酶的产生,这些胞外酶可以促进SOC的分解。随之土壤微生物的组成和结构发生变化,最终导致微生物生活史特征改变。从宏基因组数据推断出的群落特性可用于区分富营养和寡营养微生物群落。特别是,r-和K-策略分类法可能需要超越对分类基因的限制性关注,因此我们建议将该理论扩展到包括功能基因的数据集。也就是说,参与不稳定和稳定碳降解的功能基因可以分为r-和K-策略。通过这种方式,可以将草地恢复过程中微生物的表现与相关功能基因联系起来。因此,我们用土壤16S rRNA和内转录间隔区(ITS)rRNA基因测序来量化(相对)丰度,同时使用宏基因组测序来分析功能基因数据,以阐明长期草地恢复期间特殊微生物生活史特征(图S1)。我们提出以下两个假设:H1:随着草地恢复年限的增加,土壤微生物群落将从K-策略转变为r-策略;H2:与植物特性相比,土壤特性与微生物生活史特征关系更密切。
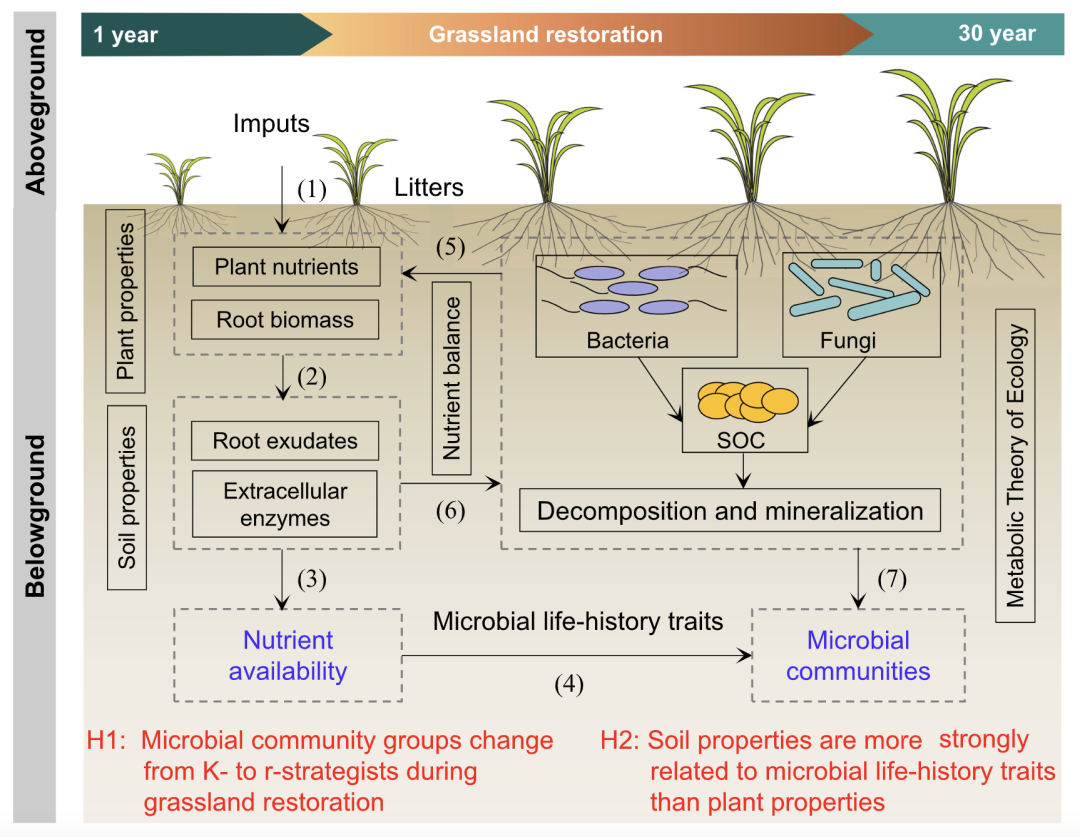
图 1. 概念框架图
概念框架图揭示了草地恢复过程中植物和土壤特性对微生物生活史特征的影响。土壤有机碳(SOC)主要由微生物对植物凋落物的转化而形成,土壤细菌和真菌是SOC分解和矿化的主要贡献者。在草地恢复过程中,植物凋落物被大量的细菌和真菌分解(1),产生各种根系分泌物和胞外酶(2),使营养物质可供微生物吸收(3),然后形成微生物群落(4)。营养元素释放和吸收的过程。大多数微生物都能获得有机养分,并将这些养分输送给植物,从而保持养分平衡(5)。反过来,营养物质又促进微生物的生长和繁殖(6-7),使它们可供植物利用,从而在草地恢复期间促进植物生长。
结 果
植被群落组成和土壤特性
在恢复早期阶段(主要是恢复1年、5年和10年),一年生草本植物(如毛细蒿、东蒿)是土壤养分、微生物生物量和酶活性较低条件下的优势物种(表1)。在恢复后期(>10年),多年生植物(如白皮针茅、茎叶蒿)是优势物种(表S1),具有较高的根系生物量、土壤养分和酶活性(表1)。叶片碳、氮含量和根系生物量随草地恢复年限增加而增加。土壤特性值,包括土壤有机碳(SOC)、水分含量(SM)、总氮(STN)、有效磷(SAP)、微生物生物量碳(MBC)、微生物生物量氮(MBN)、酸性磷酸酶(AP)、α-1,4-葡萄糖苷酶(AG)和β-N-乙酰葡糖胺苷酶(NAG),逐年增加,在草地恢复30年达到峰值,但土壤pH和微生物生物量磷(MBP)的变化趋势相反。
表1. 草地恢复对植物和土壤特性的影响(平均值±标准差)
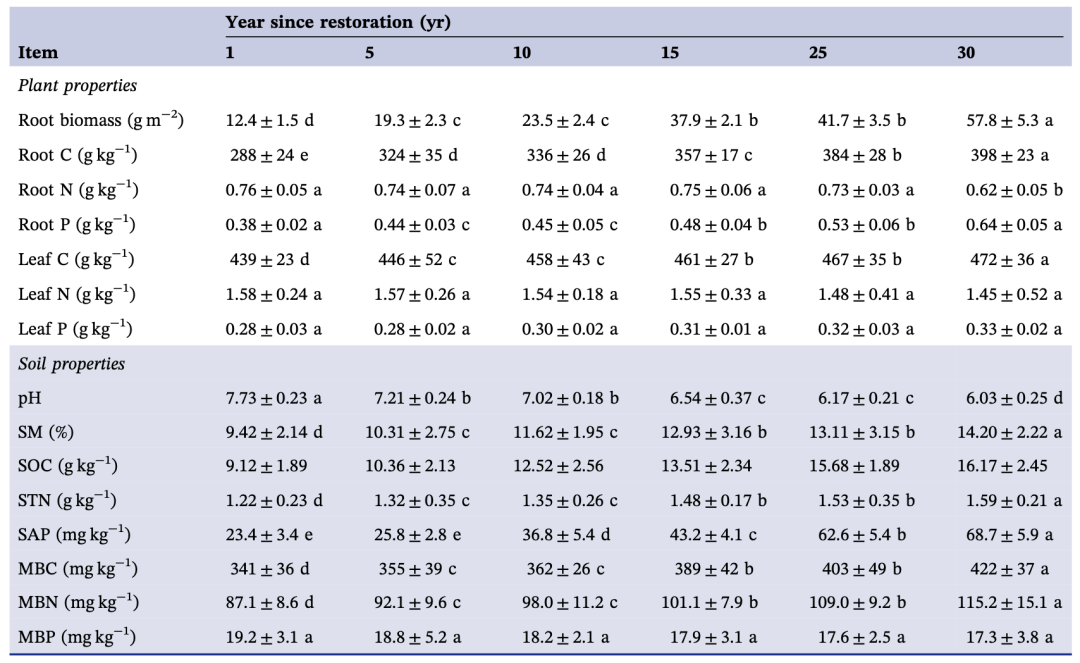
不同小写字母表示经Fisher检验后存在显著差异(P<0.05)。SM:土壤含水量;SOC:土壤有机碳;STN:土壤总氮;SAP:土壤有效磷;MBC:微生物生物量碳;MBN:微生物生物量氮;MBP:微生物生物量磷
土壤微生物群落结构
土壤细菌序列以97%的水平分为13类(表S2),优势菌(相对丰度>5%)为放线菌、酸杆菌和变形菌。土壤真菌序列以97%的水平分为8类(表S3),优势菌(相对丰度>5%)为子囊菌、担子菌和壶菌。放线菌、变形菌和拟杆菌的相对丰度随草地恢复逐年增加,在30年最高,而酸杆菌、浮霉菌和绿弯菌相对丰度在草地恢复1年时较高,随后逐年减少(表S4)。对于土壤真菌,担子菌的相对丰度在草地恢复1年时较高,随后逐年增加,而子囊菌的相对丰度逐年减少,在草地恢复30年时较高(表S5)。
细菌(表S2)和真菌(表S3)Shannon–Wiener指数在草地恢复1年时最低,随后逐年增加,在30年时达到最大值。随着序列数的增加,我们获得了平滑饱和的稀释曲线,相似度达到97%(图2A,B),细菌和真菌群落根据非度量多维标度(NMDS)显示了不同的分类(图2C,D)。真菌群落的系统发育距离比细菌群落更大(图S2)。
网络分析(图S3)表明,在草地恢复期间,所有网络中的正相关大于负相关。恢复30年的土壤微生物群落网络比其他年限更强(节点更丰富)。由于计算的模块化指数大于0.4,我们发现了典型的模块结构(表S6)。自草地恢复以来,这些经验网络中的平均聚类系数和平均路径长度随年份逐渐增加。此外模块化指数和平均连通性随年份逐渐增加,而平均路径长度和网络直径呈相反的趋势。
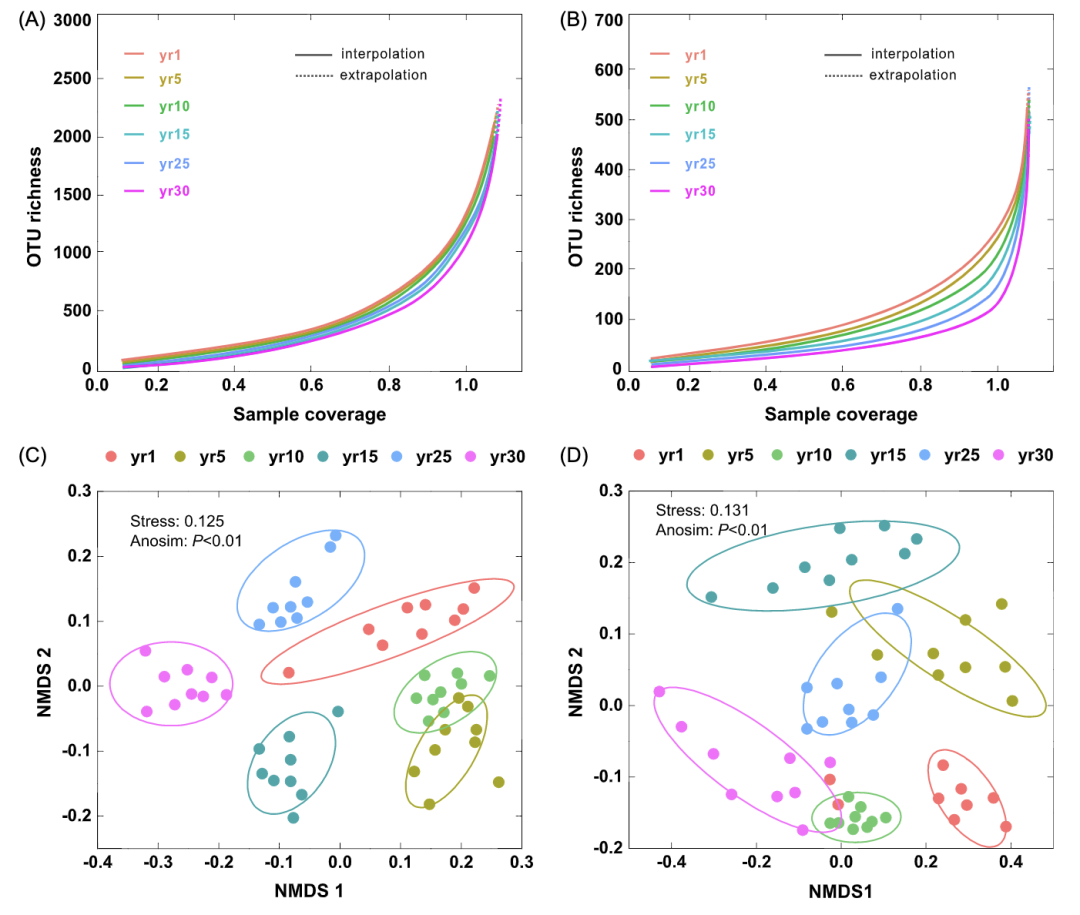
图2:草地恢复过程中细菌(A)和真菌(B)基于覆盖度的稀释(实线段)和外推(虚线段)曲线
草地恢复以来(C)土壤细菌群落和(D)真菌群落随年份的非度量多维标度(NMDS)。97%相似性的操作分类单元定义了群落,排序基于Bray–Curtis的不同性
土壤微生物生活史特征及其功能基因
自草地恢复以来,平均16S rRNA基因拷贝数、密码子使用偏性、平均基因组大小逐年增加(图3A-D),而GC含量的变化相反。恢复1年和5年的16S rRNA基因拷贝数和密码子使用偏性没有显著差异(p>0.05)。
我们分析了17个编码碳降解和固定的酶的基因家族(图3E,F)。在碳降解基因中,amyA(编码α-淀粉酶)、amyX(编码糖化酶)、abfA(编码半纤维素)和apu(编码支链淀粉)的相对丰度随草地恢复年份的增加而增加,而glx(编码降解木质素的乙二醛氧化酶)的相对丰度逐年减少(表S7)。碳固定基因(accA、aclB、acsA和rbcL)以及acsB和smtA的相对丰度逐年降低(表S8)。
真菌PLFAs逐年减少,细菌PLFAs逐年增加(图4);草地恢复对总PLFAs没有显著影响(p>0.05)。同时,自恢复以来,真菌与细菌的PLFA比、革兰氏阳性菌与革兰氏阴性菌的PLFA比和K:r比逐年降低。
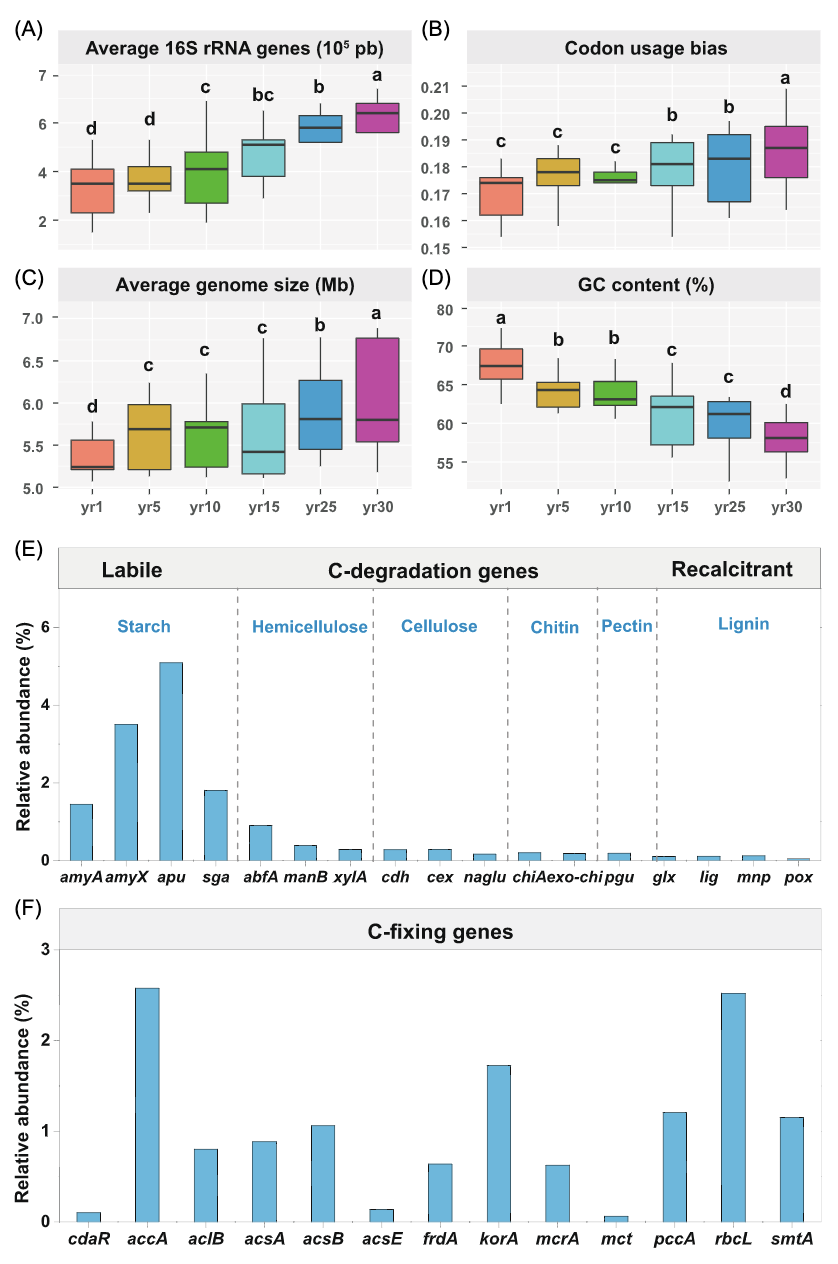
图 3. 草地恢复过程中的土壤微生物生活史特征
箱线图展示了草地恢复期间生活史特征值的变化:平均16S rRNA拷贝数(A)、密码子使用偏性(B)、平均基因组大小(C)、GC含量(D)。方框的下边界和上边界分别表示第1和第3四分位数;水平线表示平均值;竖条的上下边界分别反映了第10和第90百分位数。不同小写字母表示经Fisher检验后存在显著差异(P<0.05)。草地恢复过程中土壤微生物功能基因(E:C降解基因;F:C固定基因)的相对丰度。这些基因根据其目标底物的生物降解能力排列,从不稳定到稳定。
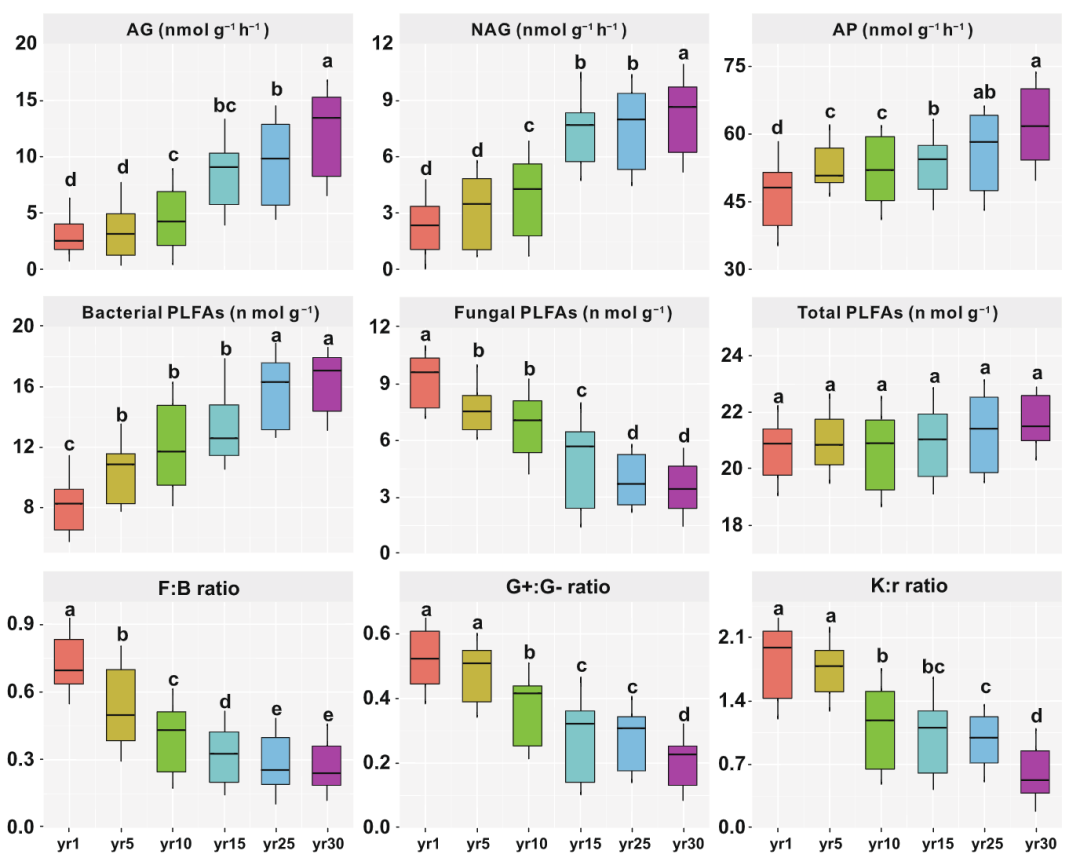
图 4. 草地恢复过程中土壤胞外酶活性和微生物群落(由PLFA表示)的变化
所有值均以平均值(标准差)表示。不同小写字母表示经Fisher检验后存在显著差异(P<0.05)。缩写:PLFA,磷脂脂肪酸;F:B比,真菌与细菌的PLFA比;G+:G-比,革兰氏阳性菌与革兰氏阴性菌的PLFA比。AG:α-1,4-葡萄糖苷酶;NAG:β-N-乙酰氨基葡萄糖苷酶;AP:酸性磷酸酶
植物和土壤特性对微生物生活史特征的重要性
酸杆菌的相对丰度与根的生物量、根的碳含量、STN、MBC、MBN、AG和NAG呈正相关(P<0.05;图5A)。相反,变形杆菌的相对丰度与土壤pH值相关,与根的生物量、根的碳含量、SOC、STN、MBC、MBN和AG呈负相关。对于土壤真菌,子囊菌的相对丰度与根的生物量、根的碳含量、STN、MBN、AG和NAG呈正相关。担子菌的相对丰度与土壤pH值呈负相关。
根据结构方程模型中的最终模型,土壤特性对微生物生活史特征有积极影响,而植物特性对微生物生活史特征则有消极影响(P<0.05)(图5B)。最重要的是,土壤特性比植物特性更好地解释了微生物生活史特征(图5B)。
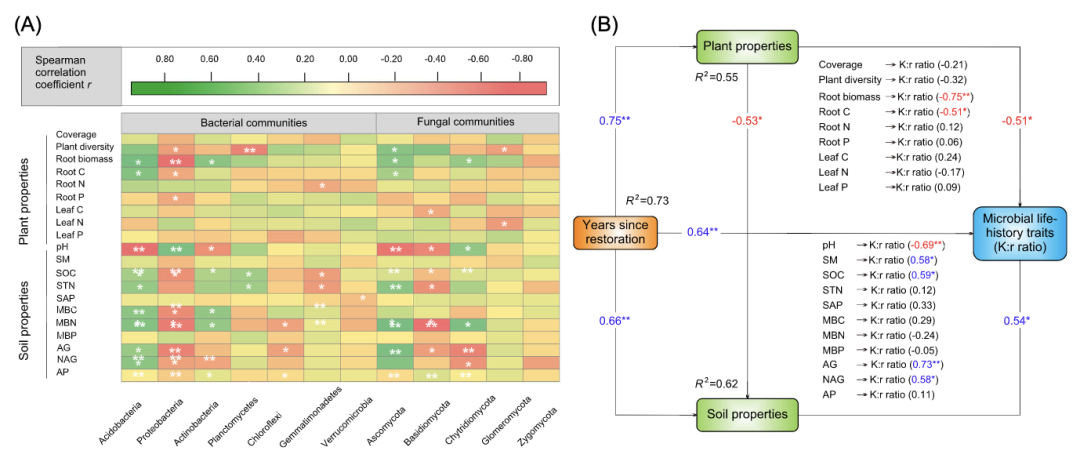
图5:优势土壤微生物(左栏,细菌群落;右栏,真菌群落)的相对丰度与植物和土壤特性之间的斯皮尔曼相关性分析
(A)。每个单元格的颜色与斯皮尔曼相关系数的值成正比。绿色表示正相关(深绿色,r=0.80);红色表示负相关(深红色,r=0.80)。* P < 0.05; ** P < 0.01. 描述植物和土壤特性对微生物生活史特征影响的最终结构方程模型(SEM)(P=0.634;ᵡ2=6.12;GFI=0.984;AIC=156.23;RSMEA=0.002,75%)(B)。矩形包括可观察变量。与箭头相邻的数字是标准化路径系数,类似于相对回归权重,并表示大小对关系的影响。R2表示变量变化的解释比例。*P<0.05;**P<0.01。红色数字表示负相关,蓝色数字表示正相关
讨 论
草地恢复对土壤微生物群落的影响
通常认为土壤细菌是r-策略菌,土壤真菌是K-策略菌。有研究表明在废弃农田的自然演替过程中,土壤真菌与细菌比显著增加,表明可能从r-策略转变为K-策略。然而,在本研究中,真菌与细菌比随草地恢复年限增加而下降(图4),在植被演替过程中,细菌可能会超过真菌,与其他有关植被演替一致。主要是因为土壤真菌,尤其是丝状真菌,比细菌在宏观尺度上更容易进入土壤孔隙。因此,r-策略细菌在草地恢复早期对不稳定碳的消耗可能受到距离的限制。随着草地恢复,这一限制解除,导致r-策略细菌增加。此外,在草地恢复过程中,革兰氏阳性菌(G+)与革兰氏阴性菌(G-)的比值也有所下降(图4)。革兰氏阴性菌比革兰氏阳性菌更喜欢利用不稳定的有机碳。K-策略真菌更适应营养不良的生态位,并能有效矿化难分解的碳,最终导致K-策略真菌减少,表明从K-策略转向r-策略,支持了我们的第一个假设。
放线菌、变形菌、拟杆菌和子囊菌通常被认为是快速生长的富营养菌,能产生较多酶用于分解新鲜有机物,并以不稳定的有机碳为食,是典型的r-策略。本研究中,放线菌、变形菌、拟杆菌和子囊菌在草地恢复30年时含量较高(表S4),并且都富含不稳定的有机碳。这些富营养微生物对营养丰富的条件反应迅速,但也能够利用蛋白质和脂质(微生物成分),并参与降解聚合物,如纤维素。然而,酸杆菌、浮游菌、绿弯菌和担子菌是K-策略菌(寡营养菌),因为它们可以在半纤维素或纤维素上生长,并矿化难分解的有机碳。这些生长缓慢或营养不足的微生物在SOC分解中发挥着重要作用,因为它们主要产生胞外酶,以在资源匮乏时降解复合物。在恢复早期阶段,低养分利用率会限制微生物的生长和活性,此时,r-策略微生物占主导地位,它们有助于干扰后的种群重建。由于土壤养分的缺乏,这些微生物在繁殖中投入比生长、代谢或提高竞争力更多的能量。然而,由于植被恢复后土壤碳输入和养分积累,这种限制可以得到缓解。在恢复后期,寡营养微生物(K-策略)类别下的担子菌门相对丰度降低。丰富的土壤养分资源改善了有机质的分解和矿化,有利于微生物生长,从而导致了高竞争力和稳定的种群(r-策略)。这种快速生长的微生物刺激根系吸收养分,然后产生更多的凋落物和根系分泌物,促进养分循环和分解之间的平衡。
在微生物网络中(图S3),恢复30年的微生物共生网络的高正相关性表明微生物群落之间潜在相互作用的比例较高。共生网络的高度模块化意味着群落可能拥有相似的生态位或模块化结构。与恢复1年相比,恢复30年的微生物群落网络更模块化,具有更复杂的群落结构(图S3)。一般认为越复杂的群落拥有越大的稳定性。因此,当应对不断变化的环境时,恢复30年的复杂微生物群落网络具有更高的稳定性。这是因为较高的土壤养分资源增加了特定微生物的代谢反应,使其在长期环境筛选后占据更多的生态位。此外,真菌显示出比细菌更高的Bray-Curtis距离(图2),表明在草地恢复期间,土壤真菌比细菌更容易变异和进化。这一发现与其他关于次生演替的研究一致,其中真菌可能在演替过程中与细菌竞争,并产生更大的变异和进化,因为它们与植物建立了密切的联系,可以更有效地利用植物中的碳。
草地恢复对土壤微生物功能基因和生活史特征的影响
由于每个门内巨大的系统发育和生理多样性,导致整个门不太可能保持一致的生态角色。例如,在变形杆菌中,β-变形杆菌表现出富营养特性(r-策略);而α-变形杆菌可能不属于r-策略菌。类似地,放线菌中的一些微生物(例如放线菌目)可能能够分解复杂且难降解的碳化合物(如木质素、纤维素),因此,表现出K-策略倾向。例如,在天然草地和森林生态系统中,放线菌生长缓慢,可以在营养不良的条件下生存,因此通常被归类为K-策略菌。相反,在增加营养的实验中,放线菌与增加的氮呈正相关,被归类为r-策略菌。除了计算寡营养生物或富营养生物的比例外,加权平均的rRNA操纵子拷贝数是微生物群落生态策略的更好反应指标。可以通过DNA数据库中的环境基因组序列方便地估计rRNA操纵子拷贝数,因为rRNA操纵子拷贝数在系统发育中是保守的。研究表明,基因组特征,如密码子使用偏性指数和rRNA操纵子拷贝数,可以代表微生物生活史策略,因为富营养生物(r-策略)通常具有较高的rRNA基因拷贝数,而寡营养生物(K-策略)通常具有较低的rRNA基因拷贝数。自草地恢复以来,rRNA基因的平均拷贝数逐年增加(图4),丰富的营养资源增加了核糖体含量,促进了富营养微生物的生长,进一步支持了r-策略菌在草地恢复过程中的主导地位。此外,微生物表现出核糖体基因的高密码子使用偏性,表明在草地恢复期间快速生长的基因表达较多。在恢复初期,土壤微生物通常比生长、代谢或增强竞争力使用更多的能量来进行繁殖(K-策略),而在恢复后期,微生物繁殖率低,存活率高,导致竞争力高,种群数量稳定。
本研究将r/K选择概念扩展到功能基因。有研究发现,植被演替增加了土壤微生物群落的多样性,以及与碳固定、碳分解(amyA、nplT、xylA、CDH和glx)相关的土壤微生物基因的丰度。植被演替增加了碳的输入,进而提升了难分解碳的基因丰度。我们的结果表明,不稳定碳基因(amyA、amyX、apu、sga)的丰度在恢复后期更高(表S7),而难分解碳基因(chiA、pgu、glx)的丰度在恢复早期更高(图S8)。这意味着土壤微生物群落的繁殖率低,但存活率高,导致在恢复后期具有较强的竞争力和稳定的K-策略种群,而土壤微生物在恢复后期的繁殖上花费的精力往往多于生长、代谢和提高竞争力(r-策略)。众所周知,微生物的功能基因可以调节土壤酶活性,胞外酶是土壤碳循环的直接驱动因素。我们发现碳固定基因丰度较高有利于土壤固碳。此外,土壤微生物通常是碳限制的,与我们先前的研究一致,由于植物产量和土壤碳含量增加,碳的有效性增强,可能有助于增加微生物生物量,形成稳定的碳,有利于SOC积累。
草地恢复对K:r比值的影响及其对微生物生活史特征的生态学意义
K:r比值与植物特性的负相关性以及与土壤特性的正相关性(P<0.05,图5B)支持了我们的第二个假设。因此,我们的数据表明,对比植物特性,微生物生活史特征的变化与土壤特性的相关性更强,类似于Zhang等人和Cui等人的发现。这是因为在草地恢复过程中,土壤特性(包括微生物生物量、养分含量和胞外酶活性)得到了改善,大大增强了微生物代谢,促进了微生物生长。然而,Mitchell等人发现,在北方生态系统中,植物群落的组成比土壤化学性质更能预测微生物群落。同样,Peay等人指出,真菌群落的组成与热带森林中植物群落的变化密切相关。这些可能是生态系统特有的。因为不同生态系统中微生物的营养需求(来自植物和土壤)差异很大。例如,在大多数草原生态系统中,养分需求主要来自植物残体,但在大多数森林生态系统中微生物残体是微生物的主要营养资源。
我们建立了一个框架,说明植物和土壤特性如何在草地恢复期间驱动土壤微生物生活史特征的变化(图6)。这种生活史框架能够直接将微生物性能与营养资源有效性联系起来。结果表明,草地恢复可以促进植物生长,增加根系生物量(表1),然后产生大量的分泌物和有机物质,提供微生物在微生境中生长。在这种情况下,营养资源的改善也可能改变具有不同生长策略的微生物的相对丰度。本研究证明,从宏基因组数据推断出的多重生态信息特征可以促进富营养-寡养框架内微生物群落基于性状的表征(图6)。然而,值得注意的是,由于细菌在土壤DNA库中的优势,宏基因组方法偏向于细菌。土壤生物地球化学过程中的其他重要介质,如真菌、土壤病毒和抗生素,没有考虑进来。因此,未来的研究应增加样本量,更好地整合土壤真核生物、病毒和抗生素,更准确地针对特定的生物地球化学过程,这对于更好地理解土壤群落、基因和过程之间的关系至关重要。
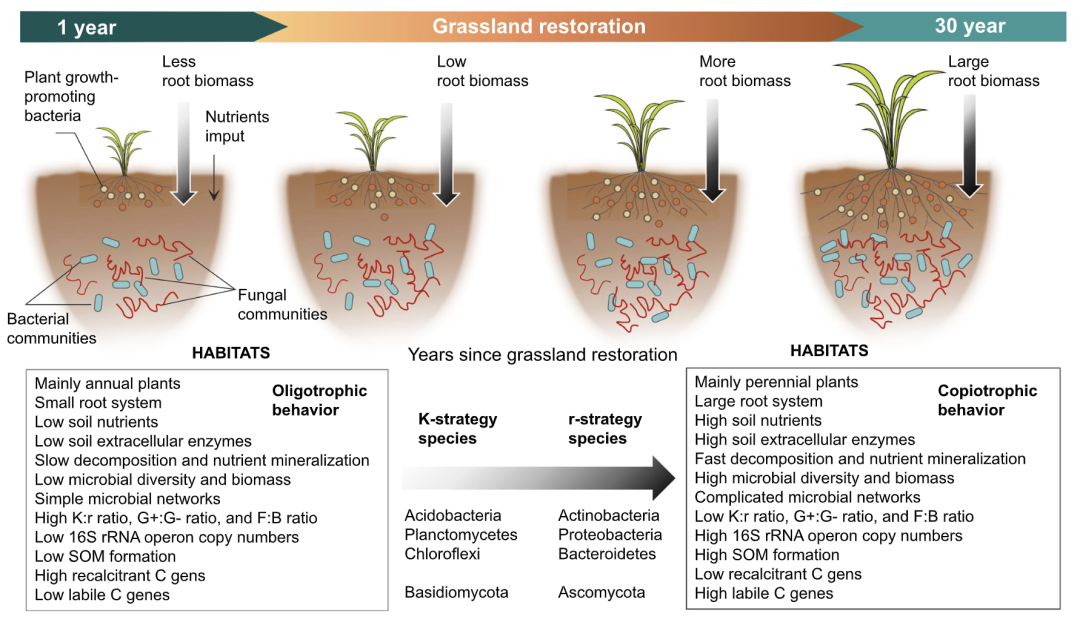
图6:揭示植物和土壤特性影响土壤微生物生活史特征的框架图
根据莫诺方程和资源有效性(含量和负荷率),r-策略微生物(富营养微生物或机会物种)被描述为生活在资源丰富的环境中,其特征是单位细胞质量的营养需求高、生长速度快和底物亲和力较弱,它们在资源丰富时优先使用易获得的土壤有机碳(例如根系分泌物和其他低分子物质或水溶性有机碳源)。相反,K -策略微生物 (寡营养微生物或平衡物种) 生长速度慢、底物亲和力较强。在草地恢复过程中,r-策略微生物逐渐增加,以提高群落生产力和机会,而K-策略微生物则由于功能冗余的调整而减少,以提高群落效率。群落在r-和k -策略之间的平衡有利于草地恢复过程中对资源扰动的适应。
结 论
从扩增子测序、宏基因组和GeoChip数据推断出的多重生态信息特征可用于研究富营养-寡营养框架内的微生物,土壤微生物生活史特征随植物和土壤特性的变化而变化。草地恢复过程中,土壤微生物由寡营养向富营养转变,与土壤特性的关系比与植物特性的关系更密切。因此,我们进一步认为,宏观生态学理论可以应用于土壤微生物生活史特性研究,并强调了植物性状(根系生物量和碳含量)和土壤性状(主要是土壤pH、SOC和胞外酶) 在草地恢复中对微生物策略改变发挥的关键作用。
方 法
研究地点
本研究在黄土高原海拔1800 - 2100米的恢复草地上进行(图S1)。该区气候温和,以温带草原为主。年降水量400-450毫米,60-75%的降水发生在7 - 9月。年蒸发量1017 - 1739 mm,年平均气温7.0℃左右,年平均日照时间约2500 h。
过度放牧和收割干草,以及耕作会导致草地退化。为保护草地资源,我国采取了多种措施(圈地和禁牧)。20世纪80年代以后,为恢复植被,该区禁止放牧,并开展了长期自然恢复试验。草地保护前,相同自然条件和土壤类型下,土壤理化性质无显著差异。随机选取一系列退化程度相似的草地,每隔几年进行一次自然修复,形成一个30年无放牧和人为干扰的恢复草地。该地区野生植物约300种,如白莲蒿, 长芒草, 鸡冠草, 冰草, 兴安胡枝子, 百里香。目前,该区是中国科学院草地动态监测的试验地
样品采集
在每个样方中,剪掉所有的枝,然后挖出所有的根(1 × 1 m2),深度50 cm。在75°C下冲洗、干燥植物样品,直到恒重,得到生物量。然后,通过0.5 mm的筛网对样品进行筛分,分析碳、氮和磷的含量。同时确定了每个样方的植被组成(表S1)。
用元素分析仪(AIC100,荷兰)分析根和叶的碳、氮含量。用70% HNO3在微波消化系统(NAI-WB,上海,中国)中消化样品12小时;用钼蓝比色法测定叶片和根系磷含量。
植物特性
TCGA样本的DNA甲基化数据(Methylation450K)是从Xena获得的,只有映射到铜死亡调节子启动子区域的探针才被用于后续分析。对于含有多个探针的基因,使用所有探针的平均β值作为甲基化水平。在差异甲基化分析中只保留了至少有5对配对的肿瘤-正常组织的16种肿瘤,并使用与差异mRNA表达分析相同的方法计算倍数变化和p值。随后,在整合了甲基化和铜死亡调控子的基因表达数据后,使用Spearman相关性分析确定了相关系数和p值。
结合铜死亡调节子的甲基化数据和临床信息后,基于Cox回归分析计算调节子甲基化的危险比。危险比 > 1代表高甲基化与不良OS相关,表示高风险。根据每个调节子甲基化水平的中位数,将每种肿瘤类型分为两组,并进行log-rank检验计算p值。
土壤特性
使用WET传感器(WET-2传感器,Delta-T Devices Ltd.,剑桥, 英国)测定土壤水分含量,使用pH计(PHS-2型,INESA仪器,上海,中国)测定土壤pH值。土壤全氮(STN)测定采用凯氏定氮法,土壤有机碳(SOC)测定采用重铬酸钾外加热法。0.5 mol L-1 NaHCO3处理后用钼蓝比色法测定土壤有效磷含量。
为测量土壤微生物生物量碳、氮、磷 (MBC, MBN, MBP, mg kg−1),先用0.5 M K2SO4提取约4.0 g新鲜制备的土壤样品,在室温下用振动筛处理1.5小时,然后用滤纸过滤(3 hw, Sartorius Stedim Biotech,哥廷根,德国)。采用熏蒸萃取法测定MBC、MBN、MBP。
MBC以EC / kEC计算,其中EC =(熏蒸土壤中提取的有机碳) -(非熏蒸土壤中提取的有机碳), kEC = 0.45
MBN以EN / kEN计算,其中EN =(熏蒸土壤中提取的总氮)-(非熏蒸土壤中提取的总氮),kEN = 0.54
MBP以EP / kEP计算,其中EP =(熏蒸土壤中提取的总磷)-(非熏蒸土壤中提取的总磷),kEP = 0.40
扩增子测序
用土壤DNA试剂盒(200)(MoBio Laboratories)提取均质土壤(0.5 g)基因组DNA。采用聚合酶链式反应(PCR)法对土壤细菌中16S rRNA基因的V3-V4区进行引物扩增,引物包括F515 (5'-GTGCCAGCMGCCGCGGTAA-3')和R907 (5'-CCGTCAATTCMTTTRAGTTT-3')。对于土壤真菌,其内转录间隔区(ITS)被用作通用DNA条形码标记,用引物ITS1F (5'-GGAAGTAAAAGTCGTAACAAGG-3')和ITS2R (5'-GCTGCGTTCTTCATCGATGC-3')扩增。将纯化的扩增子归一化后,以等摩尔含量整合,在Illumina MiSeq平台(中国杭州谷河信息技术有限公司)上进行双端测序(2 × 300 bp)。
序列数据处理
使用Quantitative Insights Into microbiology Ecology (QIIME, version 2.0, https://qiime2.org/),对序列读取执行额外的修剪和多路解编,然后对读取深度进行校正。对于FastQ文件,用Sickle软件过滤原始序列,并删除< 150bp或平均质量评分<20的序列。然后,将剩下的序列剪成ITS或16S rRNA基因序列,根据显著相似性,使用BLAST (http://sundarlab.ucdavis.edu/rice/blast/blast.html)在线搜索,选出具有代表性的序列。然后,利用QIIME将ITS或16S rRNA的条形码基因序列注释到不同的文库中,获得操作分类单元(operational taxonomic unit, OTUs),通过Vsearch (v. 1.11.1, https://github.com/torognes/vsearch/releases)筛选,进行复制、聚类和嵌合体检测。利用Greengene 13.8 (http://www.greengene.com/)软件,根据OTUs的代表性序列进行分类。每个样本的序列数由序列的大小和抽样覆盖率归一化。然后,我们使用“iNEXT”包在R统计软件中进行基于覆盖率的稀释。利用QIIME对土壤微生物多样性指标Shannon-Wiener、Simpson和Chao1进行测定。细菌和真菌的系统发育树之间的UniFrac距离使用邻居连接法进行构建(图S2)。
微生物磷脂脂肪酸(PLFAs)
土壤微生物磷脂脂肪酸(PLFAs)从6克冻干土壤样品中提取,使用氯仿-甲醇萃取混合物,加入磷酸盐缓冲液进行改性。每种脂肪酸的丰度以每克干土的纳摩尔计算。标准命名法被用来描述PLFAs。根据19:0(壬酸甲酯,C20H40O2)内部标准含量计算PLFA含量。总体而言,以PLFAs为代表的微生物群落组成分为细菌(革兰氏阳性菌和革兰氏阴性菌)和真菌,具体见表S10。G+:G-和F:B分别代表革兰氏阳性菌与革兰氏阴性菌生物量之比和真菌与细菌生物量之比。
此外,K-和r-微生物群落及功能基因的微生物生活史策略如表S11所示。用革兰氏阳性菌和革兰氏阴性菌计算K:r比。
GeoChip数据和功能基因丰度
为了确定功能基因的组成,利用GeoChip 5.0 (60 K)技术得到土壤微生物功能基因的相对丰度。基因阵列中约有6万个寡核苷酸探针,分属约400个与生物地球化学循环相关的基因家族,如碳和氮循环基因。简单地说,用Cy-3染料(GE Healthcare UK Limited)标记0.8 μg DNA,然后用QIAquick试剂盒(Qiagen)纯化,并在SpeedVac (ThermoSavant)中干燥。标记样品在67℃下杂交24小时后,利用NimbleGen MS 200微阵列扫描仪(Roche)扫描GeoChip微阵列。然后使用安捷伦数据提取软件处理图像数据和转换信号强度。剔除信号强度<2倍背景,信噪比<2的点。此外,在一个重复内测量的探针被消除。根据不同样本的相对丰度将其信号强度归一化,然后再进行生物统计分析。对函数注释的丰度进行过滤,去除丰度最小值为10%的注释。最后,通过对数变换对标注丰度进行归一化处理。
宏基因组数据与微生物生活史特征
对于宏基因组测序,使用FastDNA SPIN Kit™(MP Biomedicals, Solon, OH, USA)提取土壤基因组DNA,并根据制造商指南使用DNeasy PowerClean Pro Cleanup Kit (Qiagen, Hilden, Germany)进行纯化。将所有样品的DNA含量在PCR水中进行平衡(MoBio Laboratories, Carlsbad, CA, USA),然后将样品放在50 μL管中,并将其置于M220聚焦超声仪(Covaris, USA)中90 s,将DNA切成300 bp片段。随后,DNA样本被送至微生态科技有限公司(深圳,中国),用Illumina HiSeq4000平台(Illumina Inc., San Diego, CA, USA)按照特定的协议(www.illumina.com)准备文库和鸟枪宏基因组测序。分别使用子系统和GenBank数据库对功能基因和分类进行注释。注释过滤的最小同一性为80%,e值截止值为1 × 10−5,最小对齐长度为20 bp,这比默认参数更具限制性。
基于宏基因组测序数据计算样本特性,以阐明微生物的生活史特性。首先,根据每个OTU与已知rRNA操纵子拷贝数最近的亲缘关系,通过rrnDB数据库估计每个OTU的rRNA操纵子拷贝数。计算每个样本OTU的加权平均rRNA操纵子拷贝数,取操纵子拷贝数和每个OTU的相对丰度的乘积,并将样本中所有OTU的这一值相加。第二,假设基因组数量为单拷贝基因的平均覆盖率。用碱基对和基因组数量的商来计算基因组的平均大小。第三,计算平均基因和上调基因之间的密码子使用偏性。简单地说,ΔENC'被视为作用于上调基因密码子使用偏性的选择强度的经验估计量。对于每个基因组,测量所有编码序列(ΔENC'all)和核糖体蛋白基因(ΔENC'rib)含量的ΔENC'值,以及平均编码核苷酸频率。
然后,ΔENC'被表示为:
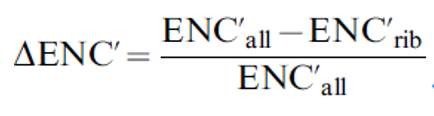
对于所有的基因组,使用EMBOSS函数getorf获得至少450 bp的开放阅读框(ORF)。对于上调的基因组,根据与已有测序基因组数据库中核糖体蛋白的相似性(e-value < 10-5)获得核糖体蛋白。最后,从NCBI基因组数据库中获取基因组鸟嘌呤胞嘧啶(GC)含量,并计算经质量筛选的reads的GC方差,如Barberán等所述。
统计分析
用R软件v. 3.4.2 (http://www.datavis.ca/R/)进行统计分析,在95% (P < 0.05)和99% (P < 0.01)概率水平上采用单因素方差分析结合Fisher检验。采用基于Bray-Curtis不相似度的非度量多维尺度法(NMDS)确定土壤微生物群落结构的变化。NMDS分析使用“vegan”包来可视化不同组之间的样本关系。不同的网络建设方法可能导致不同的信息。在一系列测试方法中,SparCC性能最高,本研究也将用其构建共现网络。利用psych R包中的“corr.test”功能,通过斯皮尔曼相关性为微生物群落构建共现网络。选择草地恢复过程中各微生物类群(细菌或真菌)中最丰富的500个OTUs构建SparCC共现网络。以r > 0.75和误发现率< 0.05为阈值对斯皮尔曼相关性和SparCC结果进行过滤。使用“igraph”R包生成网络图,共现网络中的节点代表OTUs,而边对应节点之间的显著相关性。为了更好地量化网络拓扑结构,我们计算了一组网络参数,包括节点数、边数、平均路径长度、网络直径、累积度分布、聚类系数和模块化等,并使用Gephi 0.9.2 (https://gephi.org)交互平台对网络进行可视化(图S3)。
计算斯皮尔曼系数(使用“cor”函数)以确定植物和土壤性质等环境因素与主要微生物门相对丰度的相关性,然后使用“pheatmap”包制作热图。结构方程模型(SEM)在大尺度相关性研究中比较适用,因为它允许在多个变量之间划分因果影响。扫描电镜的第一步需要建立一个基于已知影响微生物生活史性状(K:r比)的先验模型。基于植物和土壤特性对微生物生活史特征的影响(K:r比),建立了一个先验模型(图S4)。该模型包括两组:植物特性(包括植物覆盖度、多样性、根系生物量、根系C、N、P和叶片C、N、P)和土壤特性(包括土壤pH、SOC、STN、SAP、MBC、MBN、MBP、AG、NAG、AP)。在建模之前需要进行一些数据操作,以提高数据的正态性和线性。例如,根据需要对数据进行转换(包括植物覆盖度、多样性、根系生物量、根系C、N、P和叶片C、N、P、土壤pH、SOC、STN SAP、MBC、MBN、MBP、AG、NAG、AP),以满足正态性和方差齐性的假设,并认为在P⩽0.05时检验显著。将植物和土壤特性作为一个复合变量。复合变量的使用不会改变潜在的SEM模型,而是将多个概念相关变量的影响分解为一个单一的复合效应,有助于解释模型结果。在获得满意的模型拟合后,在模型中引入复合变量。由于引入的一些变量不是正态分布,路径系数不等于零的概率使用bootstrap重采样进行了测试。在这些情况下,自举比经典的最大似然估计更受青睐,因为在自举中,概率评估不是基于数据遵循特定理论分布的假设。因此,对数据进行随机抽样并进行替换,以得到与样本中发现的数据分布在经验上相关的标准误差估计。当这些数据操作完成后,我们使用数据集对模型进行参数化,并测试其整体拟合度。我们采用低卡方(χ2)值,0.05≤p≤1.00,0≤RMSEA≤0.05(接近均方根误差),比较拟合指数(CFI > 0.90)。此外,由于一些变量不正常,我们使用Bollen-Stine bootstrap检验(当0.10 < bootstrap p≤1.00时模型拟合良好)来确认模型的拟合,最终我们的模型在所有标准下都达到了可接受的拟合。有了良好的模型拟合,我们可以自由地解释模型的路径系数及其相关的P值。SEM分析使用AMOS 21.0 (SPSS Inc.,芝加哥,伊利诺伊州,美国,https://spssau.com/)进行。
引文格式:
Yang Yang, Yanxing Dou, Baorong Wang, Zhijing Xue, Yunqiang Wang, Shaoshan An , Scott X. Chang. 2022. Deciphering factors driving soil microbial life-history strategies in restored grasslands. iMeta e66. https://doi.org/10.1002/imt2.66
作者简介

杨阳(第一作者)
● 中国科学院地球环境研究所,副研究员
●2019年毕业于西北农林科技大学,获理学博士学位;获得全国林业十佳毕业生、全国王栋草业科学奖、国家奖学金等。主要从事黄土高原土壤微生态和全球变化生态学等研究,入选中国科学院地球环境研究所“青年BR”计划,中国科学院“青促会会员”,中国土壤学会青年工作委员会创新委员,先后主持国家级、省部级和中科院基金5项。以第一作者身份发表论文30余篇,包括Global Change Biology、Soil Biology and Biochemistry、iMeta等国际主流期刊,副主编著作2部,参编著作1部,担任iMeta和Carbon Neutrality期刊青年编委

王云强(通讯作者)
● 中国科学院地球环境研究所研究员,博士生导师
●研究方向为黄土高原土壤水文过程,国家WR计划青年拔尖人才入选者,国家自然科学基金优秀青年基金项目获得者,现为黄土与第四纪地质国家重点实验室副主任、黄土高原地球关键带与地表通量国家站副站长、生态环境研究室主任。一直扎根黄土高原,在黄土高原不同尺度土壤性质时空变异特征、土壤水文与植被生态互馈关系、自然和人类活动影响下黄土关键带水文过程等方面取得一定创新性成果。目前,主持国家自然科学基金4项、中科院“西部之光”创新交叉团队项目等,发表学术论文80余篇,论文总引用4000余次,3篇第一作者SCI论文入选美国ESI高被引论文

安韶山(通讯作者)
● 西北农林科技大学水土保持研究所研究员,博士生导师
●研究方向为黄土高原土壤微生物多样性和土壤有机碳固定,主持并完成多项国家自然科学基金、西部之光、国家科技支撑等相关课题。黄土高原土壤侵蚀与旱地农业国家重点实验室水土过程首席科学家,多年来聚焦黄土高原脆弱生态系统植被恢复模式下的生态学与土壤学的前沿问题,在植被恢复模式下的土壤微生物固碳及其对地上植被的响应和协同机制等方面取得了一批创新性成果。担任《应用生态学报》和《水土保持通报》编委,以第一/通讯作者身份在Global Change Biology、Soil Biology and Biochemistry、Biology and Fertility of Soils、Geoderma等国际主流杂志论文100余篇,论文总引用3000余次,入选2021年度全球高被引学者
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









