






点击蓝字 关注我们
与健康人相比,结直肠癌患者有更多环境来源的抗生素耐药基因

iMeta主页:http://www.imeta.science
研究论文
●原文:iMeta (IF 23.8)
●原文链接:
https://onlinelibrary.wiley.com/doi/full/10.1002/imt2.70008
●DOI: https://doi.org/10.1002/imt2.70008
● 2025年3月5日,香港中文大学于君教授和香港大学张彤教授等在iMeta在线发表了题为“Transmission of antimicrobial resistance genes from the environment to human gut is more pronounced in colorectal cancer patients than in healthy subjects”的文章。
● 本研究揭示了移动性ARG(mARG)从环境传播到人类肠道的途径,及其在结直肠癌(CRC)中的作用。
● 第一作者:刘伟鑫
● 通讯作者:于君(junyu@cuhk.edu.hk)、张彤(zhangt@hku.hk)
● 合作作者:刘伟鑫、刘焯晞、丁筱、殷晓乐、胡嘉麒、黄曦、沈祖尧
● 主要单位:香港中文大学李嘉诚健康科学研究所、香港中文大学深圳研究院、消化疾病研究所、医学及药物治疗学系、消化疾病国家重点实验室,中国香港特别行政区;香港大学土木工程系环境工程研究中心环境微生物组工程与生物技术实验室,中国香港特别行政区;南洋理工大学李光前医学院,新加坡。
亮 点
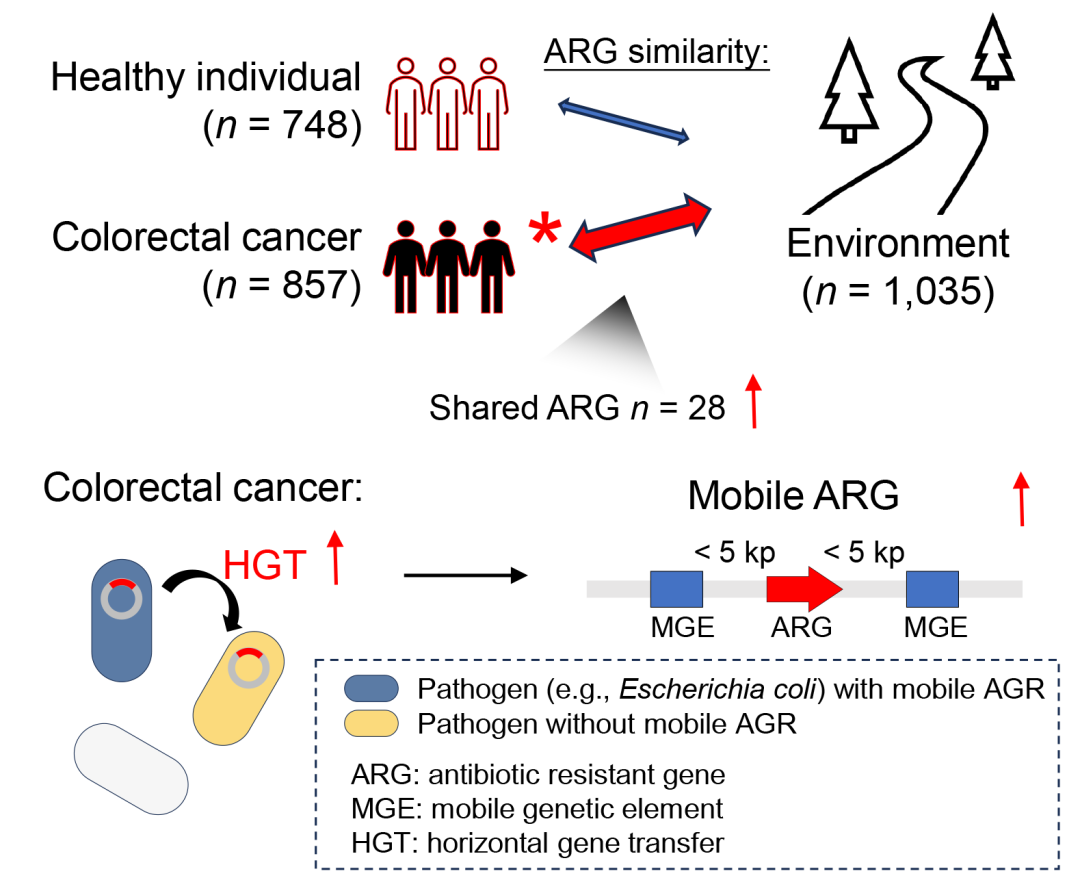
● •结直肠癌(CRC)患者相较于健康人具有更高的抗菌素耐药基因(ARG)负荷;
● 在同一城市的环境与人类肠道之间,CRC患者与环境的ARG谱比健康人更为相似;
● 与健康人相比,CRC患者拥有更多的环境-肠道共有的ARG、更多的移动性ARG(mARG),以及更高的从环境到肠道的ARG传播效率;
● mARG的细菌宿主主要为致病菌(例如大肠杆菌和共生梭菌),这些致病菌在CRC患者中相较于健康人显著富集。
摘 要
抗生素耐药性是全球主要的健康问题之一。然而,肠道耐药性的来源仍未解决。我们旨在分析环境抗生素耐药基因(ARG)对结直肠癌(CRC)的贡献。本研收集了1,605个人类粪便样本(CRC患者 = 748;健康人 = 857)和1,035个城市匹配的环境样本的宏基因组数据,其中110个CRC患者、112个健康人和56个环境样本是新收集的。与健康人相比,CRC患者的ARG负荷明显更高(p < 0.01),如多药耐药ARGs水平增加。CRC患者肠道中的ARG也与环境ARG更相似(p < 0.001)。通过比较环境和肠道ARG,28个在环境中存在的ARG同时也被鉴定为CRC富集的ARG,包括SUL2和MEXE,而健康人中未发现这些ARG。同时,与健康人相比,CRC患者中观察到更多来自环境的可移动ARG(mARG)(p < 0.05)。mARG的宿主主要是致病菌(例如大肠杆菌(E. coli)和共生梭菌(C. symbiosum))。与健康人相比,环境到肠道的水平基因转移效率在CRC患者更高。与健康人相比,CRC患者中携带特定mARG(例如E. coli-SUL2和C. symbiosum-SUL2)的致病菌丰度显著增加(p < 0.05)。因此,我们揭示了ARG从环境传播到CRC患者肠道的途径。
视频解读
Bilibili:https://www.bilibili.com/video/BV1UoQEYMEcH/
Youtube:https://youtu.be/36LUcWF8iys
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
抗菌素耐药性(Antimicrobial resistance,AMR)是全球最受关注的公共卫生问题之一。抗生素耐药基因(ARGs)在人类病原体中的广泛传播带来了突出的临床挑战,尤其是在感染管理方面。高抗菌素耐药性负担可能导致抗生素无法有效清除细菌,从而可能引发严重感染、患者预后差并增加死亡率。结直肠癌(CRC)是全球第二大致命癌症,而肠道致病菌在结直肠肿瘤的发生和发展中起着关键作用。然而,目前尚不清楚CRC患者是否具有更高的抗生素耐药携带率,以及携带ARG的肠道致病菌在CRC患者中的丰度是否增加。
人类与其生活环境密切相关且相互依存。环境微生物组的巨大多样性可以作为人类病原体获取耐药性并抵消抗生素作用的基因库。据报道,人类抗菌素耐药性的产生是由于暴露于污染环境,例如含有耐药菌的废水、受污染的食物和其他来源。更具体地说,肠道致病菌和共生菌可以通过水平基因转移(HGT)从环境来源获取ARGs,从而导致抗生素耐药性的发生发展。从机制上讲,HGT依赖于移动遗传元件(MGEs),例如细菌质粒和噬菌体,它们可以携带ARGs并促进其从环境向人类传播。因此,评估ARGs从环境到肠道的传播风险对于理解环境与人类CRC之间的联系至关重要。
在本研究中,我们收集了1,605份人类粪便样本(CRC = 748份;健康人 = 857份)和1,035份城市匹配的环境样本,并利用宏基因组测序数据进行分析(其中110份CRC样本、112份健康样本和56份环境样本为新收集的)。我们发现,与健康人相比,CRC患者的ARG负担显著更高,且其ARG谱与环境中ARG谱的相似性更高。在CRC患者中观察到比健康人更多的来自环境的移动性ARGs(mARGs)。这些mARGs的宿主主要为致病菌(例如大肠杆菌(E. coli)、肺炎克雷伯菌(K. pneumoniae)和共生梭菌(C. symbiosum))。此外,我们发现,在CRC患者中,ARGs从环境到人类肠道的传播效率增加,且携带移动性ARGs的致病菌丰度显著高于健康人。
结 果
与健康人相比,结直肠癌(CRC)患者的抗生素抗性基因(ARG)负担更高
本研究共分析了1,605例个体(748例CRC患者和857例健康人)及1,035份环境样本的鸟枪法宏基因组测序数据(图1和表S1-3)。我们首先比较了CRC患者与健康人的肠道ARG特征。对应(Correspondence analysis)分析显示,CRC患者的肠道抗性组(由整体ARG特征决定)与健康人存在显著差异(F = 3.723,p = 0.002,置换多元方差分析(PERMANOVA))(图2A,S1A)。通过16S rRNA基因水平标准化(图2B)或细菌细胞计数标准化(图S1B)测量,发现在CRC患者肠道中ARG总负荷显著高于健康人(p = 0.009,双尾Wilcoxon秩和检验)。这一结果在10个独立队列中有9个得到验证(图S1C),证实CRC与更高的ARG负担相关。
进一步评估抗生素使用对肠道抗性组的影响。本研究排除了近三个月内使用过抗生素的人。与未使用抗生素的人相比,既往使用抗生素(> 3个月前)的人肠道抗性组未发生显著改变(图S1D-E),与先前研究一致。此外,环境样本与人类肠道ARG的相似性分析表明,无论是否暴露于抗生素(> 3个月前),CRC患者的环境抗性组与肠道抗性组均呈现趋同(图S1F)。这些结果提示CRC患者的ARG负荷增加至少部分与三个月前的抗生素使用无关。
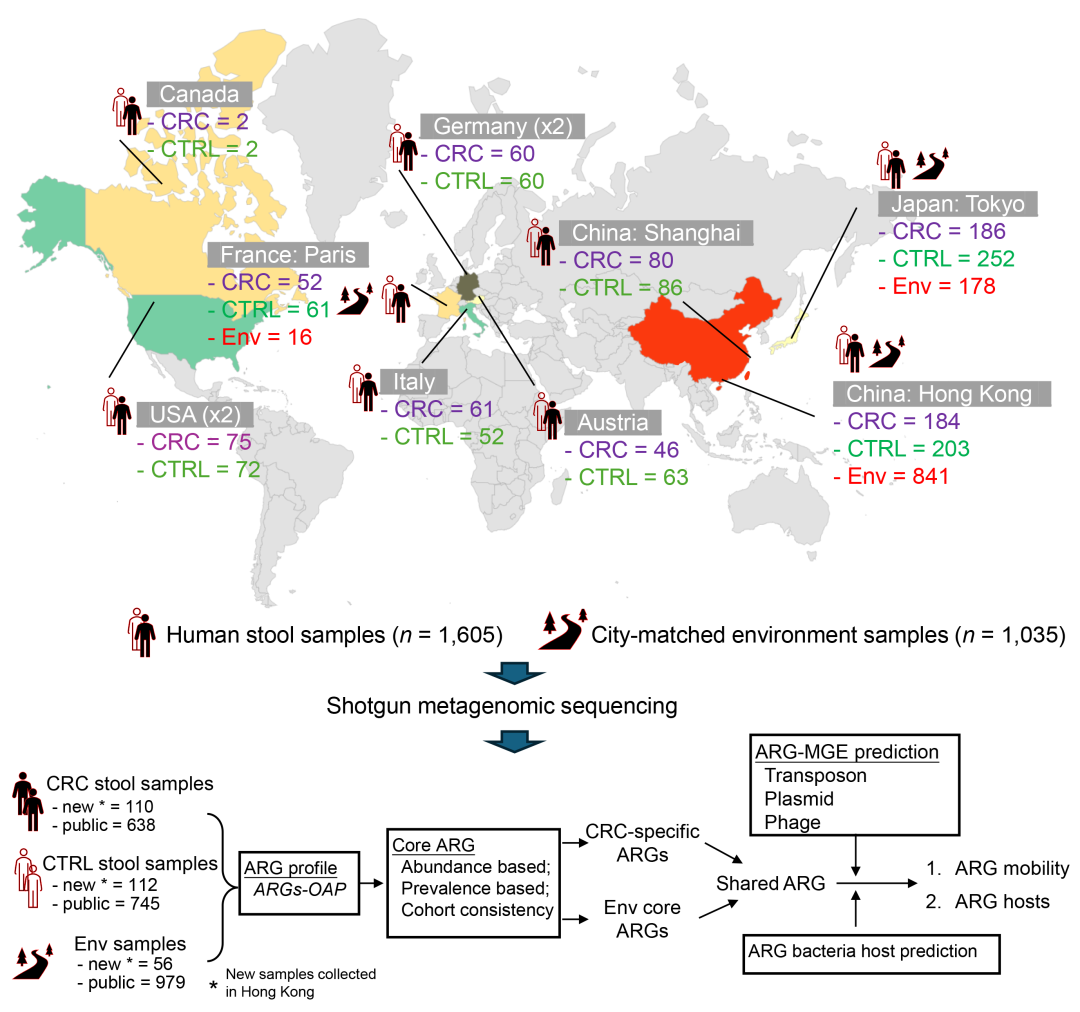
图1. 研究设计和分析工作流程
(上)在世界各地收集了结直肠癌(CRC)患者和健康人的粪便样本以及城市匹配的环境样本。(下)工作流程摘要,包括核心抗菌素耐药基因(ARG)鉴定、ARG移动性和ARG细菌宿主的分析。ARG谱通过ARGs-OAP工具获得。CTRL:健康对照;Env:环境;MGE:移动遗传元素。
CRC患者的肠道ARG特征较健康人更接近环境ARG特征
进一步探究环境ARG与人类肠道ARG的潜在关联。对应分析揭示环境ARG与肠道ARG存在显著差异(F = 439.07,p < 0.001,PERMANOVA)(图2C)。不同环境样本间的ARG特征也存在显著异质性(F = 33.06,p < 0.001,PERMANOVA)(图S1G-I)。进一步将CRC患者或健康人的粪便样本与同城市环境样本(香港、东京、巴黎)进行匹配分析(图1)。结果显示,CRC患者肠道ARG与环境ARG的差异显著小于同城市健康人(p = 2.2e-16)(图2D,S2A),表明CRC患者的ARG特征更接近环境ARG。此外,CRC患者与同城市环境样本的ARG差异显著低于跨城市环境样本(p = 1.74e-29)(图2E,S2B)。基于耐药类型特异性基因(如多重耐药ARG)的分析也观察到相似趋势(图S2C)。
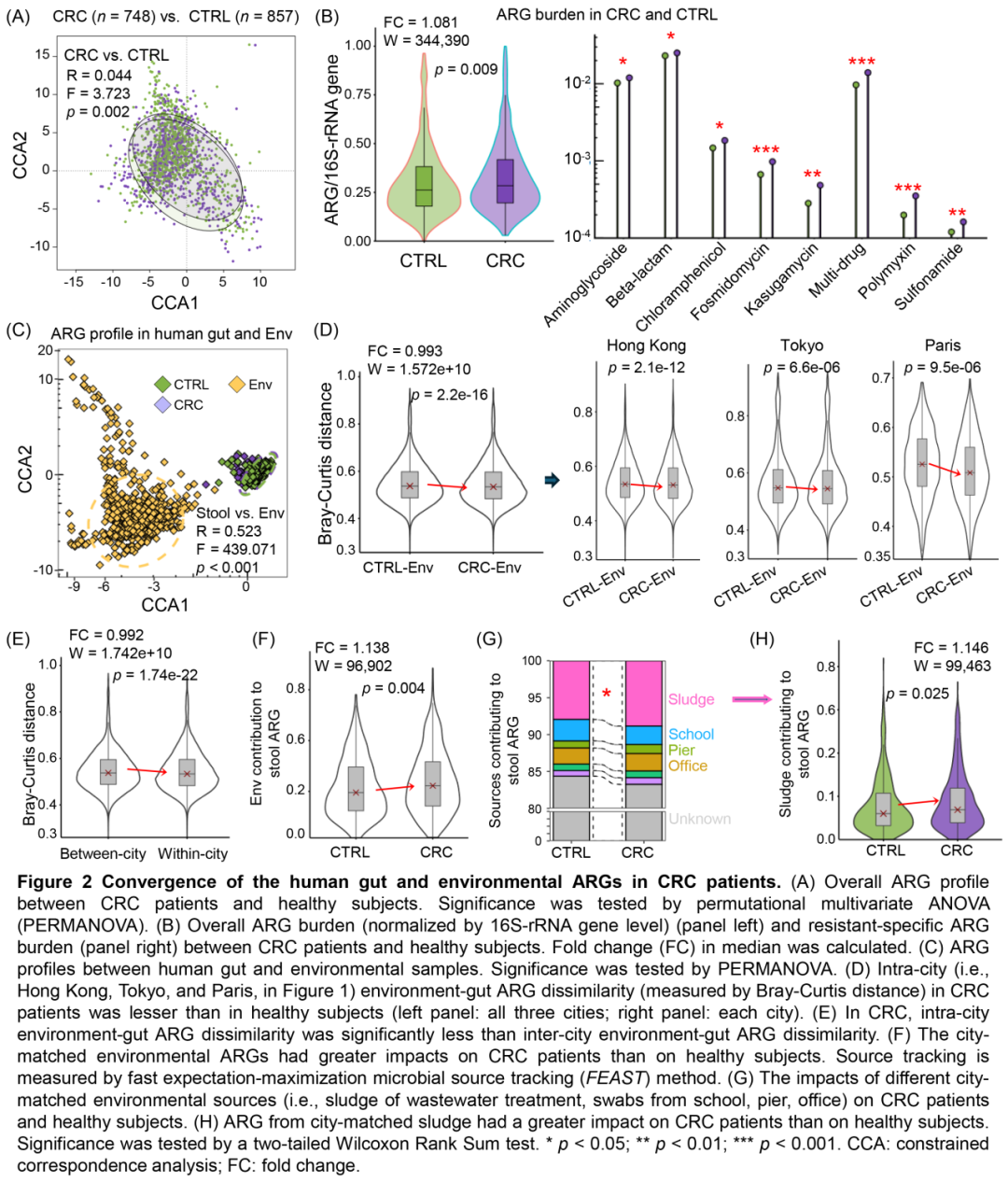
图2 . CRC患者人体肠道和环境ARG的趋同
(A)CRC患者和健康人之间的整体ARG谱。通过置换多元方差分析(PERMANOVA)检验显着性。(B)CRC患者和健康人之间的总体ARG负担(左)和耐药特异性ARG负担(右)。计算了中位数的倍数变化(FC)。(C)人类肠道和环境样本之间的ARG谱。通过PERMANOVA检验显著性。(D)CRC患者的同一城市内(即图1中的香港、东京和巴黎)环境-肠道ARG差异(通过Bray-Curtis距离测量)小于健康人(左:包括三个城市;右:每个城市)。(E)在CRC中,城市内环境-肠道ARG差异明显小于城市间环境-肠道ARG差异。(F)同一城市的环境ARG对CRC患者的影响大于对健康人的影响。来源追踪通过快速期望最大化微生物源追踪(FEAST)方法来测量。(G)不同城市匹配的环境源(即污水处理的污泥,学校、码头、办公室的拭子)对CRC患者和健康人的影响。(H)同一城市的污泥环境中的ARG对CRC患者的影响大于对健康人的影响。通过双尾Wilcoxon秩和检验来检验显著性。*p < 0.05;**p < 0.01;***p < 0.001。CCA:约束对应分析;FC:倍数变化。
通过快速期望最大化微生物来源追踪(FEAST)量化环境ARG的贡献,发现环境ARG对CRC患者肠道ARG的影响显著高于健康人(p = 0.004)(图2F,S3)。在同一城市的环境源(污水处理厂污泥、居民区/办公室/学校/码头/火车站环境拭子)中,污泥是唯一对CRC患者肠道ARG升高有显著贡献的来源(与健康人相比,p = 0.025)(图2G-H)。综上,本研究揭示了环境抗性组(尤其是污泥)对CRC患者肠道ARG负荷增加的贡献。
CRC特异性ARG与核心环境ARG存在共享
为识别CRC患者与健康人的差异ARG,我们采用两种方法(基于比值比(Meta-OR)的荟萃分析(表S4)和MMUPHin(表S5))并开发了核心指数(图S4A-C)。通过整合上述方法,共发现69个ARG在CRC患者与健康人间存在差异分布(图3A,S4D)。其中51个ARG在CRC患者中显著富集(图3A),包括与住院相关的MEXE和与磺胺类药物耐药相关的SUL2(图3B)。利用核心指数进一步筛选环境核心ARG(图S5A-B),共鉴定出140个环境核心ARG(图S5C),其中57个为污泥环境特有,其余83个来源于多种环境(表S6)。
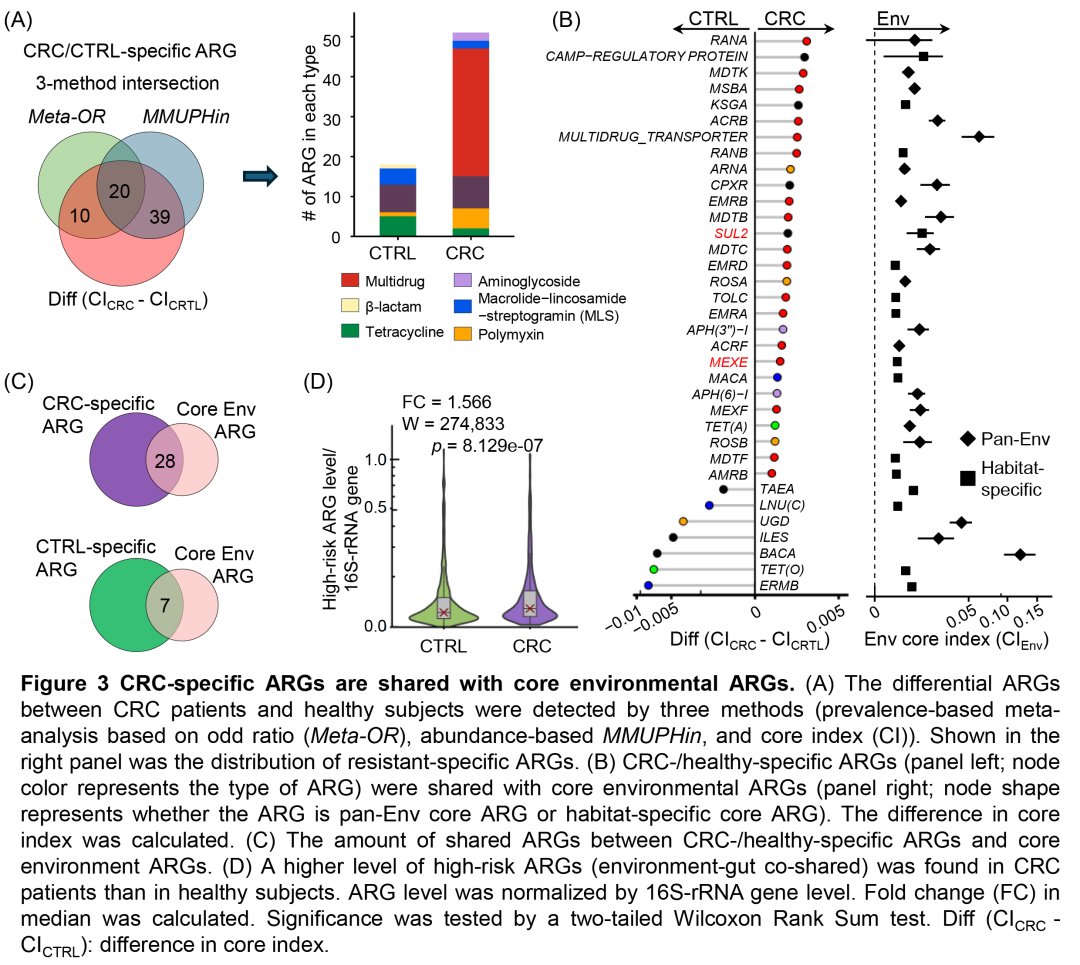
图3. CRC特异性ARG与环境核心ARG共享
(A)通过三种方法(基于比值比和基于丰度的的荟萃分析(Meta-OR和MMUPHin)和核心指数(CI))检测CRC患者和健康人之间的差异ARG。右侧显示了耐药类型特异性ARG的分布。(B)CRC/健康特异性ARG(左:节点颜色代表ARG的类型)与环境核心ARG(右:节点形状代表ARG是泛环境核心ARG还是栖息地特异性核心ARG)共享。(C)CRC/健康特异性ARG与核心环境ARG之间共享的ARG数量。(D)CRC患者的高风险ARG(环境-肠道共享)水平高于健康人。ARG水平通过16S-rRNA基因水平标准化。计算中位数的倍数变化(FC)。通过双尾Wilcoxon秩和检验检验显著性。差异(CICRC-CICTRL):核心指数差异。
通过比较环境ARG与人类肠道ARG,发现28个环境核心ARG为CRC特异性ARG(如MEXE和SUL2),7个为健康特异性ARG(如BACA)(图3B-C)。为评估ARG的健康风险,我们采用基于组学的风险分级框架,将ARG分为I-IV级风险(图S5D)。结果显示,28个环境-CRC共享ARG中,25个被纳入该框架,且这些高风险ARG在CRC患者中的丰度显著高于健康人(图3D,S5E)。环境-CRC共享ARG的存在进一步印证了CRC患者肠道ARG特征与环境的相似性。
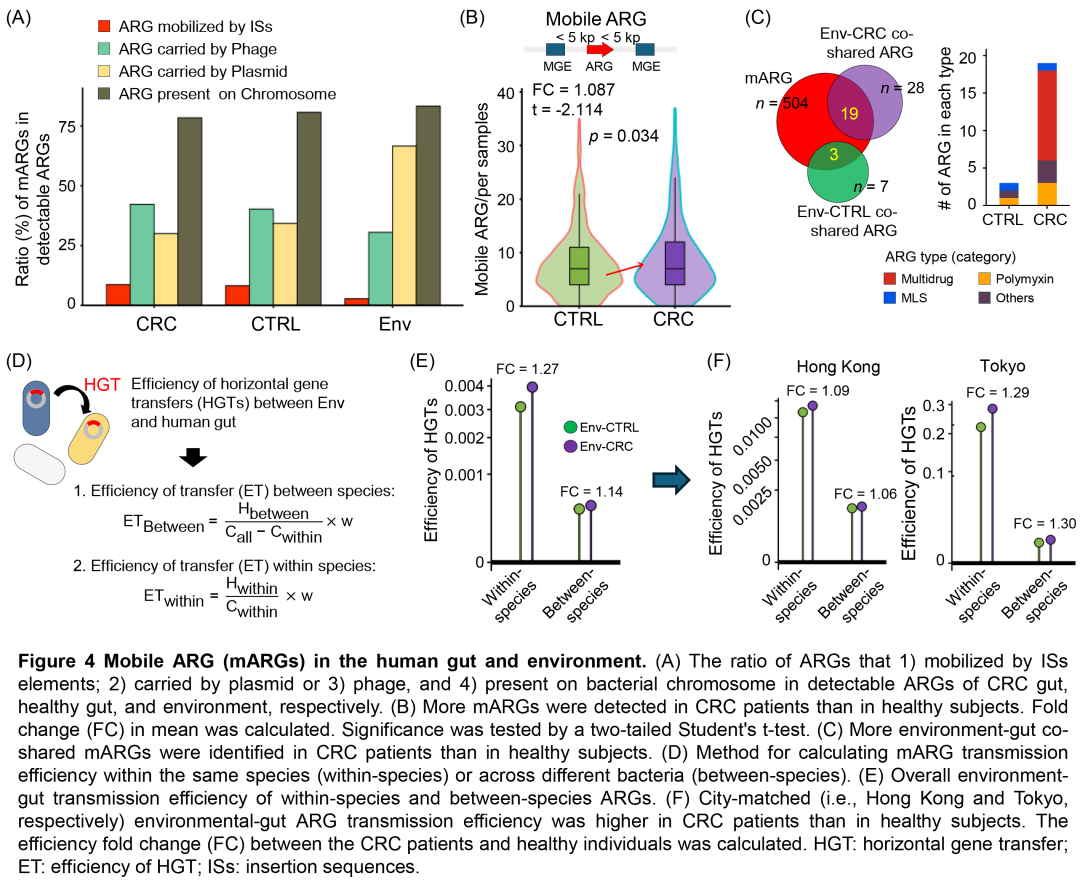
图4. 人类肠道和环境中的移动ARG(mARG)
(A)CRC肠道、健康人肠道和环境中可检测的ARG:1)由ISs元件介导的ARG;2)由质粒或3)噬菌体携带的ARG;以及4)存在于细菌染色体上的ARG的比例。(B)CRC患者比健康人检测到更多的mARG。计算平均值的倍数变化(FC)。通过双尾Student's t检验来检验显著性。(C)与健康人相比,CRC患者中鉴定出更多的环境-肠道共享mARG。(D)计算同一物种内或不同细菌之间mARG传播效率的方法。(E)物种内和物种间ARG的总体环境-肠道传播效率。(F)同一城市匹配的(即香港和东京)CRC患者的环境-肠道ARG传播效率高于健康人。计算了CRC患者和健康个体之间的效率倍数变化(FC)。HGT:水平基因转移;ET:HGT效率;ISs:插入序列。
CRC患者携带更多可移动ARG
基于环境与肠道ARG的共存现象,我们推测部分肠道抗性组可能通过水平基因转移(HGT)源自环境。为此,我们构建了整合ARG特征与多数据库的分析流程,预测ARG相关的可移动遗传元件(MGE),包括插入序列(IS)、细菌质粒及病毒/噬菌体(图S6A)。结果显示,环境ARG更多由细菌质粒携带,而肠道ARG更倾向于由噬菌体携带(图4A)。与健康人相比,CRC患者的可移动ARG显著更多(p = 0.034)(图4B,S6B),且19个环境-CRC共享ARG(包括MEXE和SUL2)被鉴定为可移动ARG(图4C,表S7)。
CRC患者的环境-肠道ARG传播效率高于健康人
抗生素抗性基因(ARG)的传播(即水平基因转移,HGT)既可发生于同种微生物内,也可跨越不同微生物。因此,我们评估了环境ARG向人类肠道在同种或跨物种微生物间的传播效率(图4D)。与预期一致,同种微生物内的ARG传播效率显著高于跨物种传播(图4E)。此外,无论环境来源如何(图S6C,表S8),环境ARG向CRC患者肠道的同种及跨物种传播效率均显著高于健康肠道(图4E)。这一现象在香港和东京(样本量充足的两个城市)中均得到验证(图4F)。综上,本研究表明CRC患者ARG负担的增加至少部分归因于环境ARG向肠道的更高传播效率。
可转移的抗生素抗性基因(mARGs)的宿主主要为机会性致病菌
微生物(尤其是病原体)是抗生素抗性基因(ARGs)的储存库,也是传播抗生素耐药性的关键载体。为此,我们通过宏基因组组装技术鉴定了环境与人类肠道中ARGs的细菌宿主(图S6A)。共构建了140,372个宏基因组组装基因组(MAGs),其中半数(n = 77,124)为物种水平的MAGs,涵盖794个不同物种(表S9、S10)。根据组装质量,物种水平的MAGs被划分为三类:高质量(n = 8,086;完整度 ≥ 90%、污染度 < 5%、菌株异质性 < 0.5)、中等质量(n = 6,565;50% ≤ 完整度 < 90%、污染度 < 5%)及低质量(n = 62,473)(图S7A、B及表S10)。我们发现高/中等质量基因组与NCBI基因组数据库的参考基因组质量相当(图S7C),因此选用这些基因组分析其携带的mARGs数量。
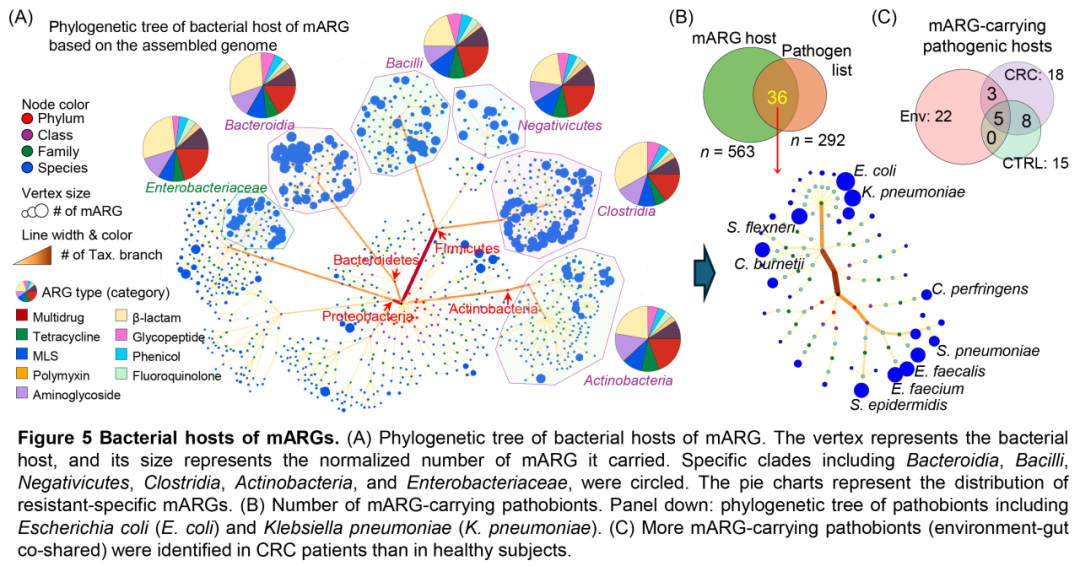
图5 . mARG的细菌宿主
(A)mARG细菌宿主的系统发育树。顶点代表细菌宿主,其大小代表其携带的mARG的标准化数量。圈出了特定的分支,包括拟杆菌、杆菌、阴性菌、梭菌、放线菌和肠杆菌科。饼图表示耐药特异性mARG的分布。(B)携带mARG的致病菌数量。下:包括大肠杆菌(E. coli)和肺炎克雷伯菌(K. pneumoniae)在内的机会致病菌的系统发育树。(C)在CRC患者中发现的携带mARG的致病菌(环境-肠道共同共享)比在健康受试者中发现的更多。
我们发现,一共可以鉴定出563个携带mARGs的细菌宿主,主要为梭菌纲(Clostridia)、芽孢杆菌纲(Bacilli)、拟杆菌门(Bacteroidetes)和γ-变形菌纲(Gammaproteobacteria,尤其是肠杆菌科(Enterobacteriaceae))(图5A)。这些细菌携带的mARGs以β-内酰胺类、多重药物及氨基糖苷类抗性ARG亚型为主(图5A、S7D)。基于病原体-宿主相互作用数据库和世界卫生组织(WHO)发布的耐药性“重点病原体”清单,我们识别出36种具有潜在致病性的mARG宿主,包括大肠杆菌(E.coli)和肺炎克雷伯菌(K.pneumoniae)(图5B)。值得注意的是,结直肠癌(CRC)患者肠道中致病性mARG宿主的数量显著高于健康人群(图5C)。此外,在所有环境样本中,污泥样本与CRC患者肠道共享的致病性mARG宿主数量最多(图S7E),提示污水与CRC可能存在正向关联。
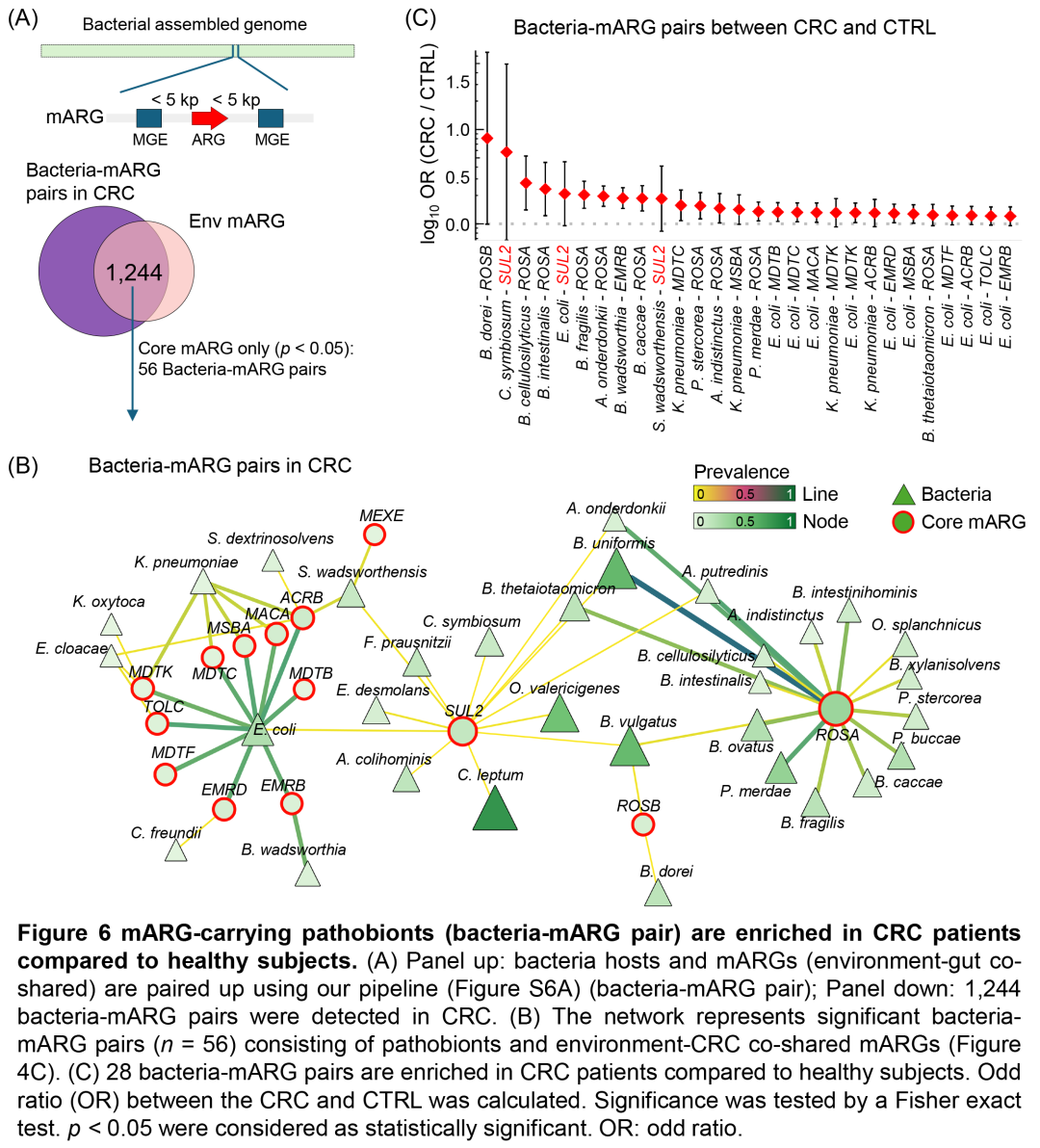
图6. 与健康受试者相比,CRC患者中携带mARG的致病菌(细菌-mARG对)更加丰富
(A)上:使用图S6A所示流程将细菌宿主和mARG(环境-肠道共享)配对(细菌-mARG对);下:在CRC中检测到1,244对细菌-mARG对。(B)网络表示由致病菌和环境-CRC共享的mARG组成的细菌-mARG对(n = 56)(图4C)。(C)与健康受试者相比,CRC患者中28对细菌-mARG对显著富集。计算了CRC和CTRL之间的比值比(OR)。通过Fisher精确检验来检验显着性。p < 0.05被认为具有统计学意义。OR:比值比。
与健康人群相比,携带mARGs的致病菌在CRC患者中显著富集
接下来,我们将细菌宿主与可移动抗生素抗性基因(mARG)配对(即细菌-mARG对),并比较了CRC患者与健康受试者中携带mARG的致病菌。在CRC患者的肠道中,我们共鉴定出1,244对细菌-mARG对,其中的mARG在环境中也存在(图6A,S8A)。在这些细菌-mARG对中,56对由致病菌(基于病原体-宿主相互作用数据库和WHO“优先病原体”列表鉴定)与环境-CRC共享的mARG组成(图4C),例如携带SUL2的大肠杆菌(E. coli-SUL2)(图6B)。与健康受试者相比,28对细菌-mARG对(如E. coli-SUL2、C. symbiosum-SUL2和Sutterella wadsworthensis-SUL2)在CRC患者中显著富集(p < 0.05,Fisher精确检验)(图6C,表S11)。同时,部分携带mARG的致病菌宿主(如大肠杆菌和肺炎克雷伯菌)的丰度在CRC患者中未发生显著变化(图S8B),这表明CRC患者中mARG水平的升高可能与细菌扩增无关。综上所述,我们的研究结果表明,携带mARG的致病菌可能从环境中传播而来,导致其在CRC患者肠道中的富集。
讨 论
肠道微生物组在结直肠癌(CRC)中的作用已得到广泛认可,但从环境中获得抗生素抗性基因(ARG)对这一恶性肿瘤的影响尚不明确。据我们所知,这是首次通过荟萃分析揭示环境ARG抗性组与CRC的关联。我们的多队列分析表明,CRC患者的ARG负担高于健康受试者。人类健康与周围环境中的微生物密切相关,微生物可以从污染的环境(如土壤、水体)中获取ARG。这些携带ARG的微生物的传播促进了人类抗生素耐药性的发展,从而降低了抗生素抗治疗感染的效果,增加了医疗负担。我们的分析显示,环境与人类肠道ARG特征在CRC患者中比在健康受试者中更为相似,这一结果通过来源追踪算法得到验证。此外,我们鉴定出28个在CRC中显著富集的核心环境ARG,而环境与健康受试者共享的ARG仅有7个。与健康受试者相比,CRC患者的ARG可移动性也有所增加。因此,我们的分析表明,环境ARG可能通过水平基因转移进入人类肠道,从而重塑CRC患者的肠道ARG特征。
我们揭示了源自环境的ARG部分通过水平基因转移(HGT)进入人类肠道。HGT高度依赖于可移动遗传元件(MGE)的存在,如插入序列(IS)、细菌质粒和噬菌体。MGE通常编码适应环境变化的特性,并作为载体在细菌细胞间传递外源DNA。同时,ARG可由MGE携带,从而实现其从环境向人类的传播。通过宏基因组组装,我们鉴定了环境中携带ARG的MGE的多样化分布,特别是发现环境ARG更多由质粒而非噬菌体携带。某些人类活动,如畜牧业、水产养殖和临床中广泛使用抗生素,对环境细菌施加了选择压力。这种压力迫使这些细菌通过MGE获得新功能,以快速适应环境变化。研究表明,质粒通常携带ARG以增强其细菌宿主的生存能力,从而对宿主适应性产生积极贡献。相反,噬菌体可能对其宿主造成显著负担:研究表明噬菌体导致细菌10%–20%的死亡率/每日,在细菌扩增期间甚至超过200%。此外,溶原性噬菌体在某些条件下(如抗生素治疗)可能被重新激活。因此,这种动态关系有助于解释环境中携带ARG的质粒更为普遍的现象。
随后,我们对环境中和人类肠道中由可移动遗传元件(MGE)携带的ARG进行了评估。我们鉴定出19个环境-CRC共享的可移动ARG(mARG),这一数量超过了环境-健康受试者共享的mARG数量(n = 3)。此外,无论环境来源如何,CRC患者的环境-人类ARG传播效率均高于健康受试者(图S6C)。我们知道,来自同一城市的健康个体和CRC患者暴露于相似的环境风险中。然而,健康个体由于肠道微生物组功能健全且免疫功能正常,对携带ARG的细菌及其他病原体的定植具有更强的抵抗力。相比之下,癌症患者由于免疫状态受损且对抗机会性病原体的能力下降,更容易受到感染。此外,癌症患者频繁就医和住院期间面临更高的细菌感染风险,从而增加了医院获得性感染的可能性。我们的分析显示,污泥样本与CRC患者肠道微生物组之间的ARG具有显著相似性。这可能是因为我们内部队列中,人类样本收集的医院与污泥样本收集的河流之间的距离非常接近。综上所述,这些发现表明,暴露于受污染的环境(如废水和医院废物)可能促进ARG向人类的传播,从而可能增加CRC患者的ARG水平。
我们的分析显示,与健康受试者相比,环境与CRC患者之间共享的致病菌更多(图4G),包括产生多种基因毒素以破坏人类宿主双链DNA的大肠杆菌,以及通过产生支链氨基酸激活CRC中Hedgehog信号通路的共生梭菌(C. symbiosum)。由大肠杆菌或共生梭菌携带的mARG(如SUL2,与磺胺类药物耐药相关,而磺胺类药物代谢物具有包括抗癌在内的多种生物活性)在CRC患者中也显著富集。另一方面,水平基因转移(HGT)可能由炎症驱动。众所周知,慢性肠道炎症是结直肠肿瘤发生的重要推动因素。炎症环境为耐氧和致病菌的增殖创造了有利条件,这些细菌特别容易参与HGT,例如先前研究和我们的研究中发现的肠杆菌科(图5A)。综上所述,我们鉴定出携带ARG的致病菌,这些致病菌可能是环境ARG向CRC患者肠道传播效率增加的原因。
本研究存在一些局限性。首先,我们的分析基于多个回顾性队列,因此主要发现是相关性研究,无法确定环境ARG与CRC肿瘤发生之间的因果关系。其次,仅有三个队列包含城市匹配的环境数据和人类CRC样本,这限制了我们关于CRC中环境ARG结果的普适性。为了增强未来研究的可靠性,收集更多城市匹配的环境和人类样本至关重要。第三,本研究中尚未确定已鉴定的携带mARG的致病菌的功能作用及其对CRC肿瘤发生的影响。
结 论
我们揭示了可移动抗生素抗性基因(mARG)从环境传播到CRC患者肠道的途径。ARG传播效率的提升导致环境mARG在肠道中的富集,从而增加了CRC患者的ARG负担。总的来说,我们的研究为环境抗性组与CRC患者之间的关联提供了新的见解。
方 法
人类粪便样本
本研究共纳入了1,605名个体,包括来自我们内部队列的748名CRC患者和857名健康受试者(n = 220),以及来自9个不同国家/城市(n = 1,382)的已发表粪便宏基因组数据集(图1和表S1、2)。来自内部CRC队列(n = 220)的粪便样本由110名CRC患者和112名健康受试者组成,他们接受了香港中文大学赛马会大肠癌教育中心的结肠镜检查。排除标准为:1)过去3个月内使用过抗生素;2)素食;3)过去3个月内接受过侵入性医疗干预;4)曾患过任何癌症或肠道炎症性疾病。与健康受试者相比,结直肠癌患者在诊断前没有发现明显的抗生素使用趋势(OR = 0.93,p > 0.05;Fisher精确检验)。所有受试者在粪便采集时结肠粘膜完整,并签署了知情同意书。临床研究方案已获得香港中文大学-新界东联网临床研究伦理委员会批准(编号CRE-2010.198和CRE-2011.297)。
此外,我们在PubMed上搜索了2014年1月至2023年1月发表的结直肠癌研究,发现了9项对人类粪便样本进行散弹枪宏基因组测序的研究。共有1,382名人类受试者,其中包括638名结直肠癌患者和745名健康受试者。样本大小和样本采集详情如表S1-2所示。宏基因组数据集从欧洲核苷酸档案馆(ENA)和日本DNA数据库下载,使用以下标识符:Zeller等人的ERP005534,Feng等人的ERP008729,Yu等人的PRJEB10878,Vogtmann等人的PRJEB1244,Hanningan等人的PRJNA389927,Wirbel等人的PRJEB27928,Thomas等人的SRP136711,Liu等人的PRJNA731589,以及Yachida等人的DRA006684和DRA008156。
环境样本
我们还收集了1,035个城市匹配的环境样本,包括来自我们内部队列的废水处理污泥(n = 56),以及从住宅(n = 344)、办公室(n = 32)、学校(n = 110)、码头(n = 174)和火车站(n = 172)获得的绒拭子(来自MetaSUB联盟(表S1、3)。2021年,我们从香港的混合移动床生物膜反应器(HMBBR;一种废水处理工艺)中收集了污泥样品。
此外,为了提高环境数据的代表性,我们收集了更多来自全球65个国家/地区的可公开获取的环境样品,包括空气(n = 81)、土壤(n = 198)、水(n = 64)、废水处理厂(WWTP,n = 303)、垃圾填埋场(n = 26)和沉积物(n = 261)(表S3和图S1G)。
宏基因组测序
分别按照制造商的说明使用QIAamp DNA Stool Mini Kit(德国Qiagen)和DNeasy PowerSoil Kit(德国Qiagen)从粪便样本和污泥样本中提取总DNA。宏基因组测序在Novogene(中国北京)使用Illumina测序平台以双端150-bp进行。
细菌抗菌素耐药基因(ARG)分析
使用默认参数的KneadData(v0.7.2)对所有宏基因组测序数据进行质量控制。此工具可以消除宿主或其他来源的污染读数并仅保留微生物读数。ARGs-OAP(v3.2.2)(默认设置)用于ARG分析。然后将识别出的ARG分类为耐药特定类型或亚型,并通过估计的细胞计数或16S rRNA基因拷贝数对测序读段(read)的计数进行标准化。细胞计数或16S rRNA基因拷贝数(即系数)是通过与必需单拷贝标记基因数据库进行映射或通过校正的16S rRNA序列的拷贝数来估计的。ARG负担定义为所有已识别ARG的标准化丰度之和。细菌分类学概况由MetaPhlAn3(v3.0.6)使用默认设置生成。
追踪人类粪便样本中ARG来源的分析
使用默认设置的R包FEAST(v0.1.0)来估计环境来源对人类粪便ARG概况的影响
核心ARG识别
我们旨在识别环境和人类肠道中的核心ARG。为此,我们制定了一个核心指数(CI),其中考虑了不同群体或环境来源之间的ARG流行率、ARG丰度和ARG一致性。第i个ARG的拟议CI计算如下:
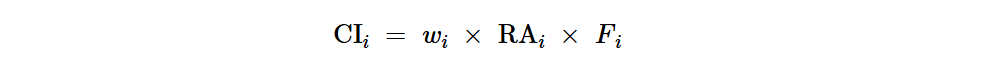
CRC患者和健康受试者之间第i个ARG的CI差异(CIdiff)定义为:
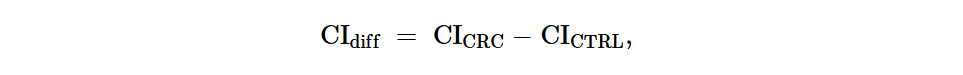
其中RAi和Fi分别是第i个ARG的相对丰度和流行率。wi是考虑第i个ARG在不同群体或环境源中是否存在的因素,即在单个群体或环境源中流行率 > 25%的ARG被认为存在于该群体或环境源中。
为了了解CI和CIdiff的重要性,我们获得了总体CI密度分布图和CIdiff密度,分布图,并根据观察到的密度形状分别对密度拟合适当的数学分布。p值由累积概率p(CI ≥ q)计算得出。显著性水平α设置为0.05。
宏基因组组装和移动遗传元件(MGE)预测
我们开发了一个流程来重建ARG相关的移动遗传元件。首先使用MEGAHIT(v1.2.8)组装宏基因组测序读段,以生成细菌基因组重叠群(contig);排除contig长度<1kb的重叠群。然后我们使用DeepARG(v1.0.1)和RGI(v5.1.0)预测重叠群上的ARG。工具DeepARG比其他工具更准确,可以显著减少假阴性;而工具RGI可以注释ARG上的单核苷酸多态性(SNP)。包括CARD、ARDB和UNIPROT在内的ARG数据库被用于ARG注释。为了预测MGE,我们首先应用BLAST(v2.9.0+)工具,使用ISfinder数据库注释携带ARG的重叠群上的插入序列(IS)或转座子。ARG也可以由质粒或噬菌体携带,从而通过接合、转座或转化促进ARG传播。因此,我们还预测了携带ARG的重叠群中存在质粒或噬菌体。PlasFlow(v1.1)用于质粒预测;而VirSorter(v1.0.6)、DeepVirFinder(v1.0)和geNomad(v1.7.1)用于噬菌体预测。我们将所有基因组信息(包括预测的ARG、ISs元素的基因组位置以及注释为质粒或噬菌体的重叠群)整合在一起,以获得ARG的移动性。
ARG细菌宿主预测
使用MaxBin(v2.0)将Contigs分成分箱组装,以获得细菌基因组。通过CheckM(v1.1.2)使用lineage_wf的预设工作流程(默认参数)评估每个组装的基因组质量。根据指南,按质量标记组装:完整性 ≥ 90、污染 < 5和菌株异质性 < 0.5的基因组为高质量(HQ);完整性 < 90、完整性 ≥ 50和污染 < 5的基因组为中等质量(MQ);其余为低质量(LQ)基因组。此外,我们从NCBI基因组数据库中检索了分离株的细菌基因组。我们发现组装基因组(具有HQ和MQ)和参考基因组之间质量相当。重建的基因组使我们能够识别细菌基因组中质粒和噬菌体的存在。系统发育树是根据细菌分类信息构建的。
ARG传输效率分析
我们使用基于已发表的方法,计算环境和人类肠道之间的ARGHGT效率,并根据样本量进行了微小修改。ARG的HGT效率是根据组装的细菌基因组和这些组装携带的移动ARG的数据计算的。具体而言,同种细菌细胞之间的转移效率(ET)计算如下:
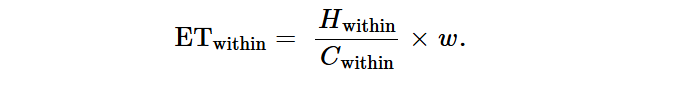
不同物种细菌细胞之间的转移效率计算如下:
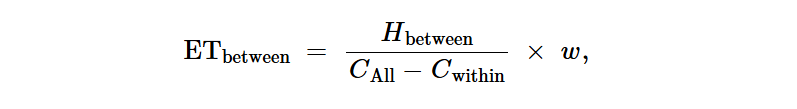
其中Hwithin和Hbetween分别代表物种内和物种间环境-肠道HGT事件的数量。w是考虑样本大小的因素,计算方法为1/(h1Xh2),其中和分别代表栖息地1和2中的样本数量。和计算方法如下:
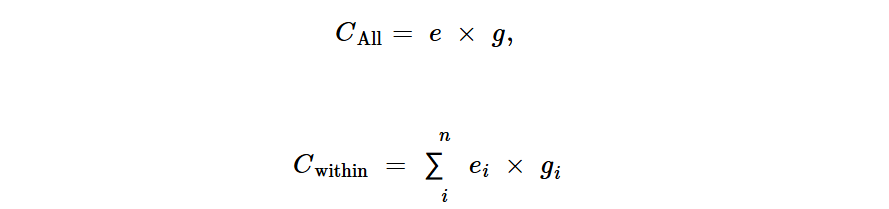
其中e和g分别是在环境和人类肠道中识别出的基因组数量。ei和gi分别表示环境和人类肠道中第i个物种的基因组数量。Cwithin中的n表示环境和人类肠道之间共享的物种数量。
统计分析
数据以均值±标准差(SD)或中位数(第一四分位数,第三四分位数)表示,视情况而定。数据通过双尾Student's t检验或双尾Mann Whitney U检验进行分析。所有分析均在开源框架R软件(v3.5.2)下进行。基于ARG丰度的荟萃分析采用MMUPHin(v1.16.0)方法完成。基于ARG流行率的荟萃分析使用metafor(v4.4-0)包进行。Fisher精确检验用于评估CRC患者中携带mARG的细菌与健康受试者相比的富集情况。所有差异在p < 0.05时被认为具有统计学显著性。为校正多重检验,p值通过Benjamini-Hochberg错误发现率(FDR)校正进行调整
代码和数据可用性
人类粪便测序数据已存放在基因组序列档案库(GSA)中,登录号为CRA022416(https://ngdc.cncb.ac.cn/gsa/search?searchTerm=CRA022416)。环境测序数据已存放在GSA中,登录号为CRA022431(https://ngdc.cncb.ac.cn/gsa/search?searchTerm=CRA022431)。使用的数据和脚本保存在GitHub(https://github.com/WilsonYangLiu/Environmental-ARGs-in-CRC-resistome.git)。补充材料(图表、表格、图形摘要、幻灯片、视频、中文翻译版和更新材料)可在网上DOI或iMeta Science(http://www.imeta.science/)中找到。
引文格式:
Weixin Liu, Harry CH Lau, Xiao Ding, Xiaole Yin, William Ka Kei Wu, Sunny Hei Wong, Joseph JY Sung, Tong Zhang*, Jun Yu*. 2025. Transmission of antimicrobial resistance genes from the environment to human gut is more pronounced in colorectal cancer patients than in healthy subjects. iMeta 4: e70008. https://doi.org/10.1002/imt2.70008
作者简介

刘伟鑫(第一作者)
●香港中文大学博士后。
●研究菌群在消化道疾病中的作用,以第一作者(含共一)在Gastroenterology、Gut、iMeta、Nature Communications等期刊发表SCI论文10篇。

于君(通讯作者)
● 香港中文大学教授,博士生导师。
● 欧洲科学院院士、香港科学院院士。

张彤
● 香港大学教授,博士生导师。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期
iMetaOmics封面

1卷1期

1卷2期

2卷1期
期刊简介

“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









