







本篇内容为scanpy的可视化方法,可以分为三部分:
- embedding的散点图;
- 用已知marker genes的聚类识别(Identification of clusters);
- 可视化基因的差异表达;
我们使用10x的PBMC数据集(包含68k个细胞)。Scanpy在其发行版中包含了这个数据集的缩减版,该数据集只包含700个细胞和765个高变基因。此数据集已经过预处理和UMAP计算。
在本篇内容里,我们使用到以下标记基因(来自于已知的文献结论,比如B-cell的标记基因为CD79A, MS4A1):
- B-cell: CD79A, MS4A1
- Plasma: IGJ (JCHAIN)
- T-cell: CD3D
- NK: GNLY, NKG7
- Myeloid: CST3, LYZ
- Monocytes: FCGR3A
- Dendritic: FCER1A
1. 降维的散点图(二维)
基于scanpy,tSNE、UMAP和其他几个embedding的散点图可以从文档轻松找到。比如可以看这里的选项列表:sc.pl.tsne,sc.pl.umap。
我们先进行初始化设置:
import scanpy as sc
import pandas as pd
from matplotlib.pyplot import rc_context
sc.set_figure_params(dpi=100, color_map = 'viridis_r')
sc.settings.verbosity = 1
sc.logging.print_header()
"""
scanpy==1.6.0 anndata==0.8.0 numpy==1.21.6 scipy==1.8.0 pandas==1.4.2 scikit-learn==1.0.2 statsmodels==0.11.0 python-igraph==0.8.0
"""
加载pbmc缩减版数据集:
pbmc = sc.datasets.pbmc68k_reduced()
# 检查pbmc内容
pbmc
"""
AnnData object with n_obs × n_vars = 700 × 765
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
这里补充关于adata的obsm,varm和obsp的内容:
- obsm:对于观测的多维注释(即对于矩阵的行的多维注释),它是可变的ndarray,长度为n_obs,维度为2至多维。这里的m指的就是multi-dim多个维度的,obs_m对应于obs,但obs的每个成员都是一维的观测注释,obs_m的每个成员(X_pac和X_umap)都是多维的观测注释。
- varm:用于描述特征的,与obsm相对应。
- obsp(obs pair):针对观测的配对的注释(存储为稀疏矩阵),稀疏矩阵两维都是n_obs,obsp通常用于描述观测与观测之间的距离和连通性。比如:
pbmc.obsp['distances']
"""
<700x700 sparse matrix of type '<class 'numpy.float64'>'
with 6300 stored elements in Compressed Sparse Row format>
"""
行和列索引 距离
(0, 9) 8.365935325622559
(0, 54) 8.560888290405273
(0, 94) 7.486799716949463
......
(699, 695) 3.6524178981781006
1.1 可视化基因表达量
对于散点图,参数color是可视化的一个值,可以是Adata的任何基因或者obs里的任何对象,注意obs是存储注释信息的dataframe。
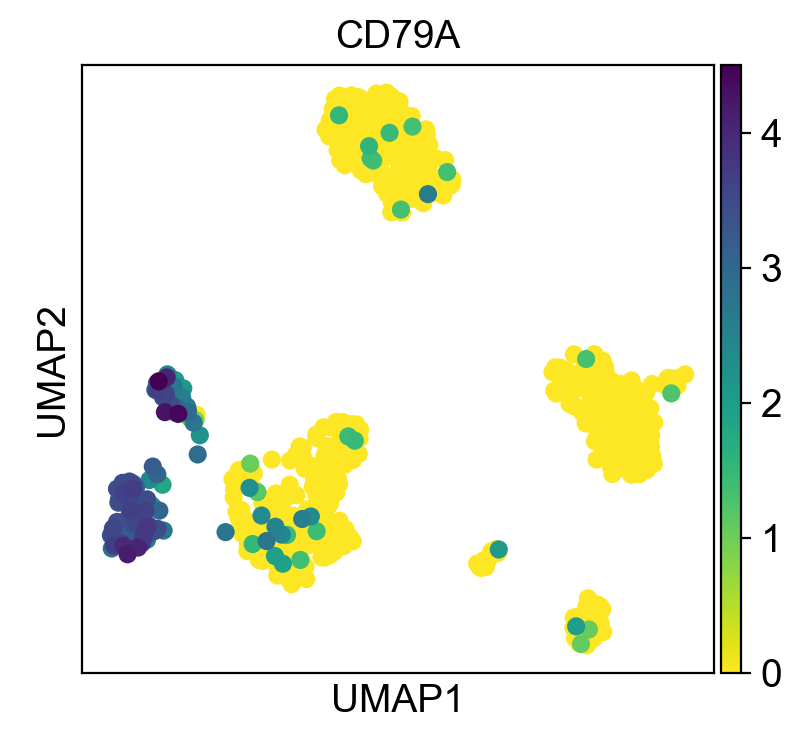
可视化基因CD79A在所有细胞中的表达量分布,由于pbmc这个adata已经有X_umap,我们可以用sc.pl.umap实现UMAP下的基因表达分布:
# rc_context用于指定figure大小
with rc_context({
'figure.figsize': (4, 4)}):
sc.pl.umap(pbmc, color='CD79A')
可以给多个值。在下面的示例中,我们将绘制6个基因:“CD79A”、“MS4A1”、“IGJ”、“CD3D”、“FCER1A”和“FCGR3A”,以了解这些marker基因的表达。
此外,我们还将绘制另外两个值:
- 一个是每个细胞的UMI计数数
n_counts(UMI检测到的基因越多,数据越复杂); - 一个是一个分类值categorical value
bulk_labels(来自10x的细胞原始标签)。
使用参数ncols控制每行的可视化案例数。可以使用vmax调整绘制颜色的最大值(同样vmin可以用于最小值)。在本例中,我们使用vmax='p99',这意味着使用99%作为最大值。如果要分别为多个可视化案例设置vmax,则vamx可以是一个数字或一组数字。
此外,我们还使用frameon=False移除可视化图的边框,并用s=50设置点大小。
ncols = 4 表示画图一行4个图。
with rc_context({
'figure.figsize': (3, 3)}):
sc.pl.umap(pbmc, color=['CD79A', 'MS4A1', 'IGJ', 'CD3D', 'FCER1A', 'FCGR3A', 'n_counts', 'bulk_labels'],
s=50, frameon=False, ncols=4, vmax='p99')
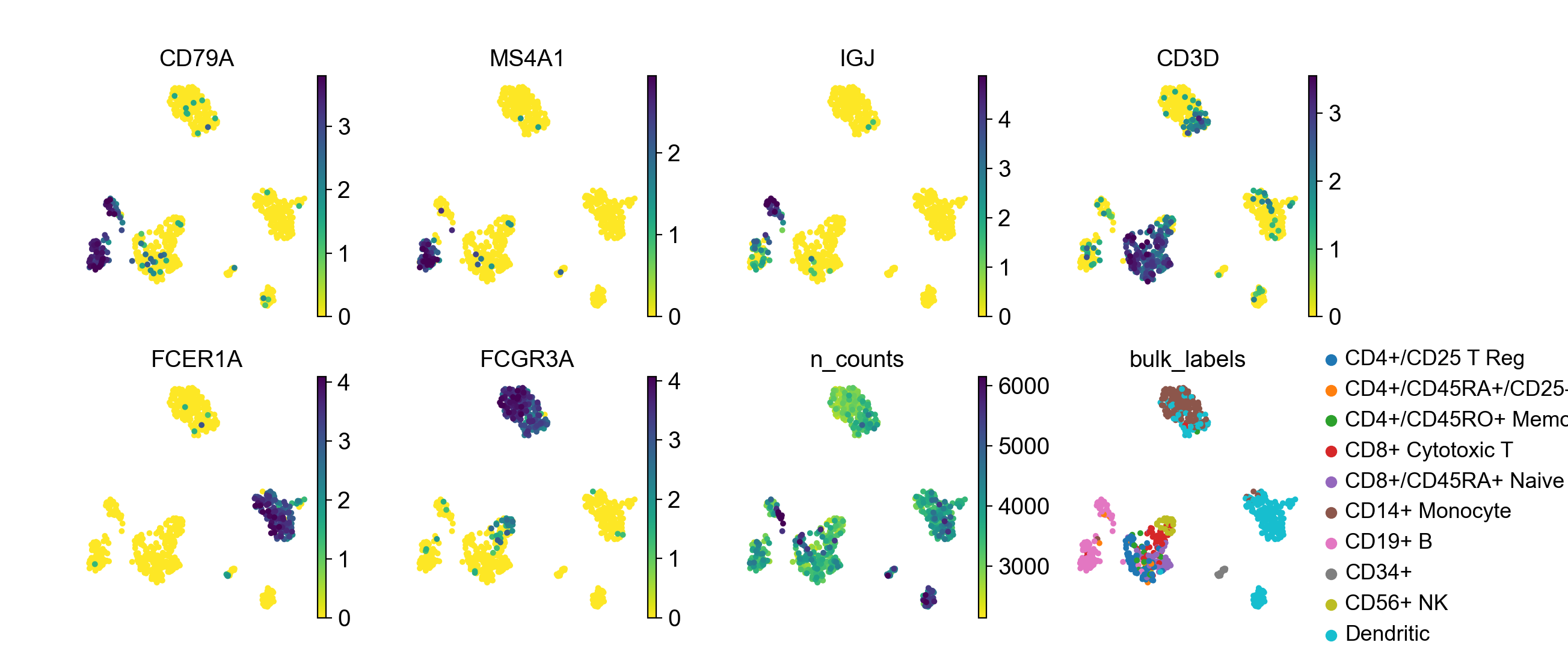
在图中,我们可以看到标记基因的细胞群与原始细胞标签的大概一致性。
散点图函数还有很多选项,可以微调图像。例如,我们可以如下所示查看clustering:
# 用leiden聚类计算, 结果存储到'clusters'中
sc.tl.leiden(pbmc, key_added='clusters', resolution=0.5)
pbmc
"""
AnnData object with n_obs × n_vars = 700 × 765
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain', 'clusters'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups', 'leiden'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
注意到,obs中多了一个注释"clusters",下一步用"clusters"作为可视化的绘制值:
with rc_context({
'figure.figsize':  最低0.47元/天 解锁文章
最低0.47元/天 解锁文章















 76
76











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









