






GO_KEGG_GSEA Enrichment
library(clusterProfiler)
library(org.Mm.eg.db) ## 人的就改成org.Hm.eg.db
DEG <- nrDEG
log2FoldChange<-DEG$log2FoldChange
logFC_cutoff <- mean(abs(log2FoldChange) ) + 2 * sd(abs(log2FoldChange))
logFC_cutoff <- round(logFC_cutoff,2)
DEG$change = as.factor( ifelse( DEG$pvalue < 0.05 & abs(DEG$log2FoldChange) > logFC_cutoff,
ifelse( DEG$log2FoldChange > logFC_cutoff , 'UP', 'DOWN' ), 'STABLE' ) )
table(DEG$change)
#### 第二步,从org.Hs.eg.db提取ENSG的ID 和GI号对应关系
keytypes(org.Mm.eg.db)
DEG$ENSEMBL <- rownames(DEG)
> df <- bitr( rownames(DEG), fromType = "ENSEMBL", toType = c( "ENTREZID" ), OrgDb = org.Mm.eg.db )
'select()' returned 1:many mapping between keys and columns
Warning message:
In bitr(rownames(DEG), fromType = "ENSEMBL", toType = c("ENTREZID"), :
11.19% of input gene IDs are fail to map...
#### 第三步,将GI号合并到DEG数据框内
DEG <- merge(DEG, df, by='ENSEMBL')
head(DEG)
dim(DEG)
#### 第四步,提取上调和下调的GI集
gene_up <- DEG[ DEG$change == 'UP', 'ENTREZID' ]
gene_down <- DEG[ DEG$change == 'DOWN', 'ENTREZID' ]
gene_diff <- c( gene_up, gene_down )
gene_all <- as.character(DEG[ ,'ENTREZID'] )
geneList <- DEG$log2FoldChange
names( geneList ) <- DEG$ENTREZID
geneList <- sort( geneList, decreasing = T )
## KEGG pathway analysis
kk.up <- enrichKEGG( gene = gene_up ,
organism = 'mmu' ,
universe = gene_all ,
pvalueCutoff = 0.05 ,
qvalueCutoff = 0.2 )
kk.down <- enrichKEGG(gene = gene_down ,
organism = 'mmu' ,
universe = gene_all ,
pvalueCutoff = 0.05 ,
qvalueCutoff = 0.2 )
library(ggplot2)
kegg_down_dt <- as.data.frame( kk.down )
kegg_up_dt <- as.data.frame( kk.up )
down_kegg <- kegg_down_dt[ kegg_down_dt$pvalue < 0.05, ]
down_kegg$group = -1
up_kegg <- kegg_up_dt[ kegg_up_dt$pvalue < 0.05, ]
up_kegg$group = 1
dat = rbind( up_kegg, down_kegg )
dat$pvalue = -log10( dat$pvalue )
dat$pvalue = dat$pvalue * dat$group
dat = dat[ order( dat$pvalue, decreasing = F ), ]
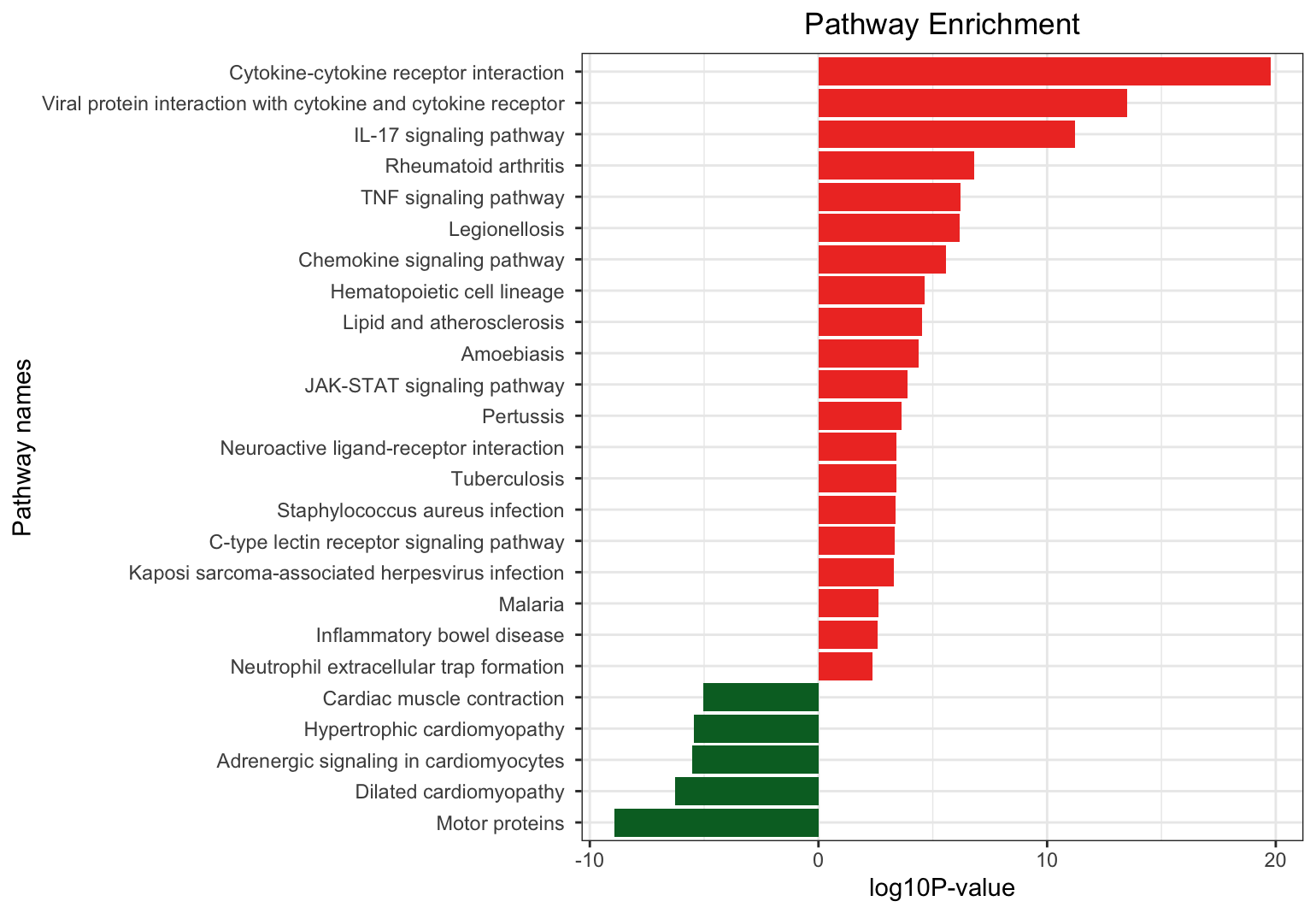
g_kegg <- ggplot( dat, aes(x = reorder( Description, order( pvalue, decreasing=F ) ), y = pvalue, fill = group)) +
geom_bar( stat = "identity" ) +
scale_fill_gradient( low = "#006D2C", high = "#EF3B2C", guide = FALSE ) +
scale_x_discrete( name = "Pathway names" ) +
scale_y_continuous( name = "log10P-value" ) +
coord_flip() + theme_bw() + theme( plot.title = element_text( hjust = 0.5 ) ) +
ggtitle( "Pathway Enrichment" )
## 这里添加对通路名称的修改,太长了总是有小鼠信息
dat$Description <- gsub(" - Mus musculus \\(house mouse\\)", " ", dat$Description)
## 在正则表达式中,括号和句点是特殊字符,需要使用双反斜杠进行转义。
## 这样可以确保准确匹配字符串 "- Mus musculus (house mouse)"。
## 这里的KEGG分析只把,满足FC阈值pvalue < 0.05
## 的上调和下调分别富集。得到BH校正后p.adj<0.05的通路 geneID
#### GO analysis
# 做GO数据集超几何分布检验分析,
# 重点在结果的可视化及生物学意义的理解。
g_list=list(gene_up=gene_up,
gene_down=gene_down,
gene_diff=gene_diff)
go_enrich_results <- lapply( g_list , function(gene) {
lapply( c('BP','MF','CC') , function(ont) {
cat(paste('Now process ',ont ))
ego <- enrichGO(gene = gene,
universe = gene_all,
OrgDb = org.Mm.eg.db,
ont = ont ,
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
readable = TRUE)
print( head(ego) )
return(ego)
})
})
}
n1 <- c('gene_up','gene_down','gene_diff')
n2 <- c('BP','MF','CC')
for (i in 1:3){
for (j in 1:3){
fn <- paste0('./Step05-GO_dotplot_',n1[i],'_',n2[j],'.png')
cat(paste0(fn,'\n'))
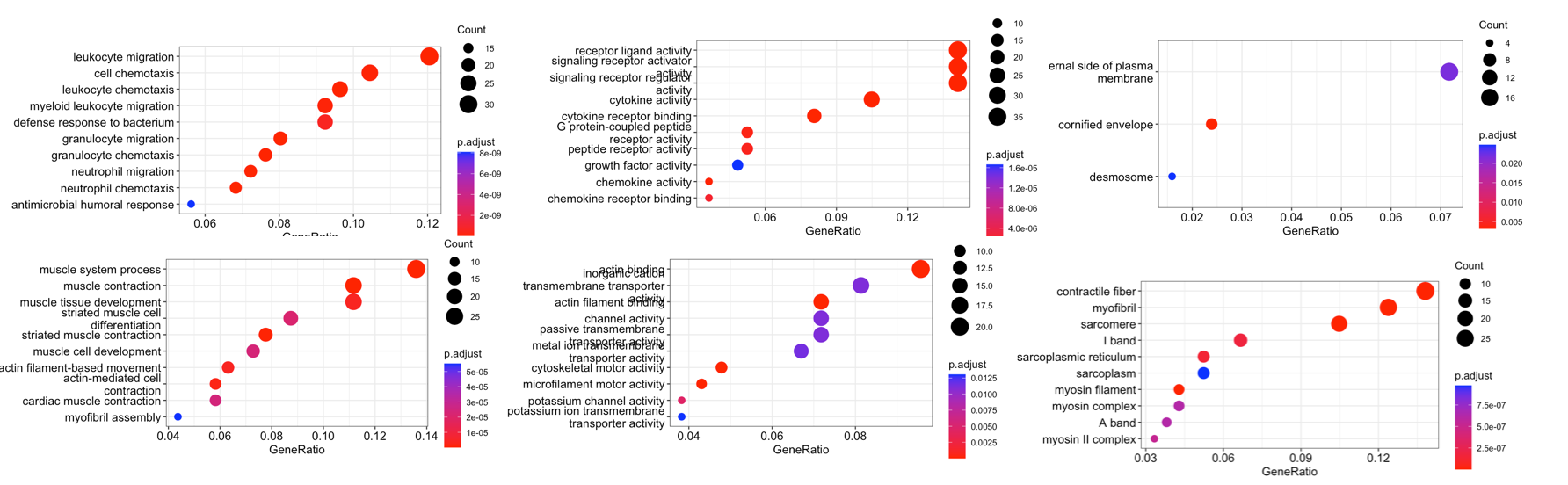
png(fn,res=150,width = 1080)
print(dotplot(go_enrich_results[[i]][[j]]))
dev.off()
}
}
## 检查一下组别上下调
sum(exprSet_focus["ENSMUSG00000000263",][1:15])
sum(exprSet_focus["ENSMUSG00000000263",][16:31])
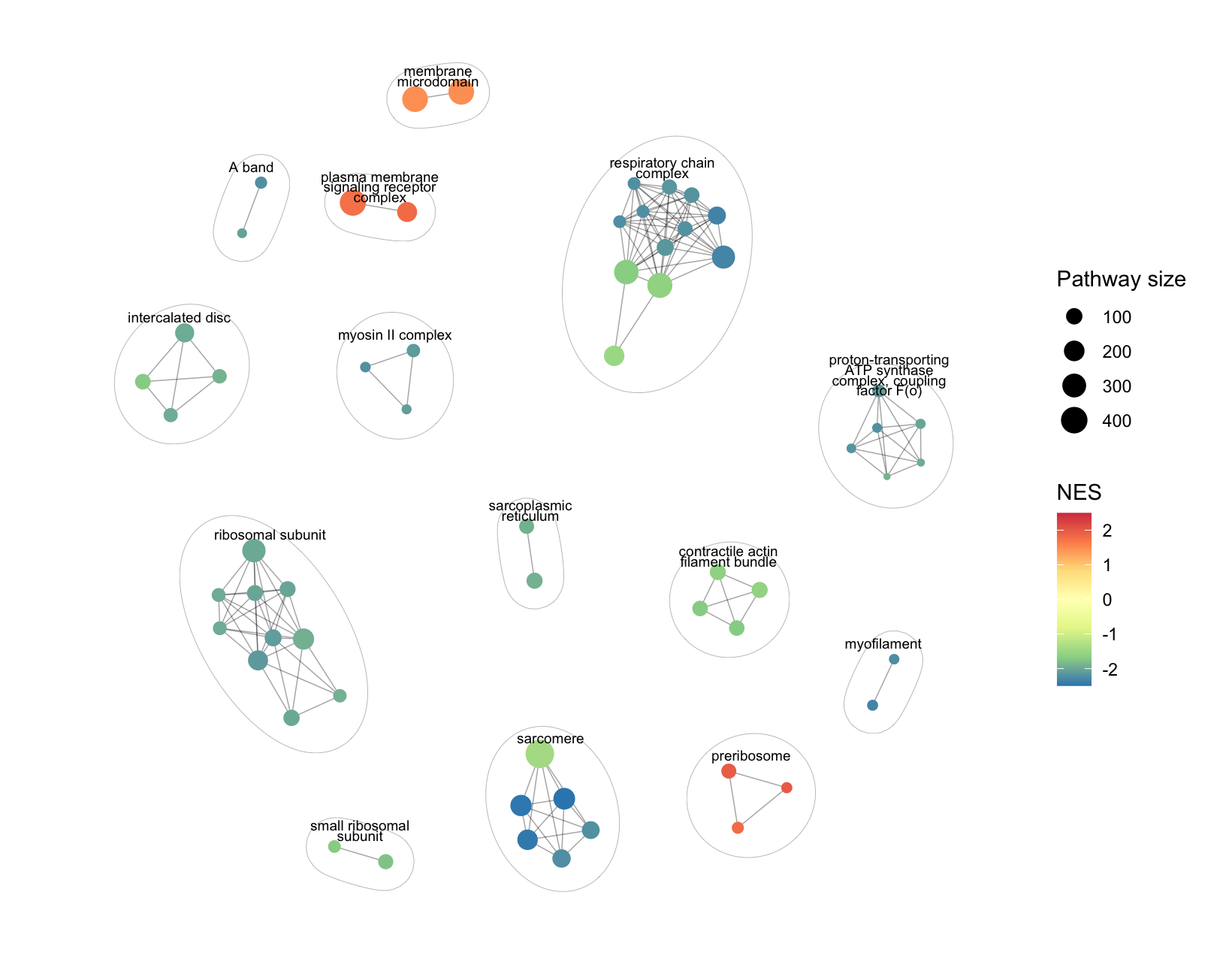
## aPEAR包绘制功能富集网络图 https://mp.weixin.qq.com/s/aUzDc9yi-U2LVvj2LUjo1w
BiocManager::Install("aPEAR")
library(aPEAR)
library(clusterProfiler)
library(org.Mm.eg.db)
library(DOSE)
library(ggplot2)
geneList <- DEG$log2FoldChange
names(geneList) <- DEG$ENTREZID
geneList <- sort(geneList,decreasing = T)
enrich <- gseGO(geneList, OrgDb = org.Hs.eg.db, ont = 'CC')
library(GSEABase)
library(clusterProfiler)
library(enrichplot)
geneList <- DEG$log2FoldChange
names(geneList) <- DEG$ENTREZID
geneList <- sort(geneList,decreasing = T)
d <- 'msigdb_v2023.2.Mm_GMTs/'
gmts <- list.files(d,pattern = '*.entrez.gmt')
gsea_results <- lapply(gmts, function(gmtfile){
# gmtfile=gmts[2]
geneset <- read.gmt(file.path(d,gmtfile))
print(paste0('Now process the ',gmtfile))
egmt <- GSEA(geneList, TERM2GENE=geneset, verbose=FALSE,eps = 1e-10,
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
head(egmt)
return(egmt)
})
## 提取GSEA结果
gsea_results_list<- lapply(gsea_results, function(x){
cat(paste(dim(x@result)),'\n')
x@result
})
gsea_results_df <- do.call(rbind, gsea_results_list)
## 写的批量生成,但是图片太多,不适合全部输出,从结果表格里选择性画出来会好一些
for (i in 1:19){
for (j in 1:nrow(gsea_results[[i]])){
fn <- paste0('./GSEA_',i,'_',j,'.png')
cat(paste0(fn,'\n'))
png(fn,res=150,width = 1080,height = 800)
print(gseaplot(gsea_results[[i]],geneSetID = j,title=gsea_results[[i]][j]$Description,
color.line="#FC9272",color="#810F7C",color.vline="#9EBCDA"))
dev.off()
}
}
## 比如我在这里,选择关心的和心脏相关的,这里需要找一下目标通路在第几基因集
## 可以从gsea_results_df输出后看出现的行数,再根据gsea_results_list,每个集合的结果加起来
gseaplot(gsea_results[[11]], "GOBP_MEMBRANE_DEPOLARIZATION_DURING_CARDIAC_MUSCLE_CELL_ACTION_POTENTIAL")
## 查看某个集合下的前20个通路
dotplot(gsea_results[[5]], showCategory=20)
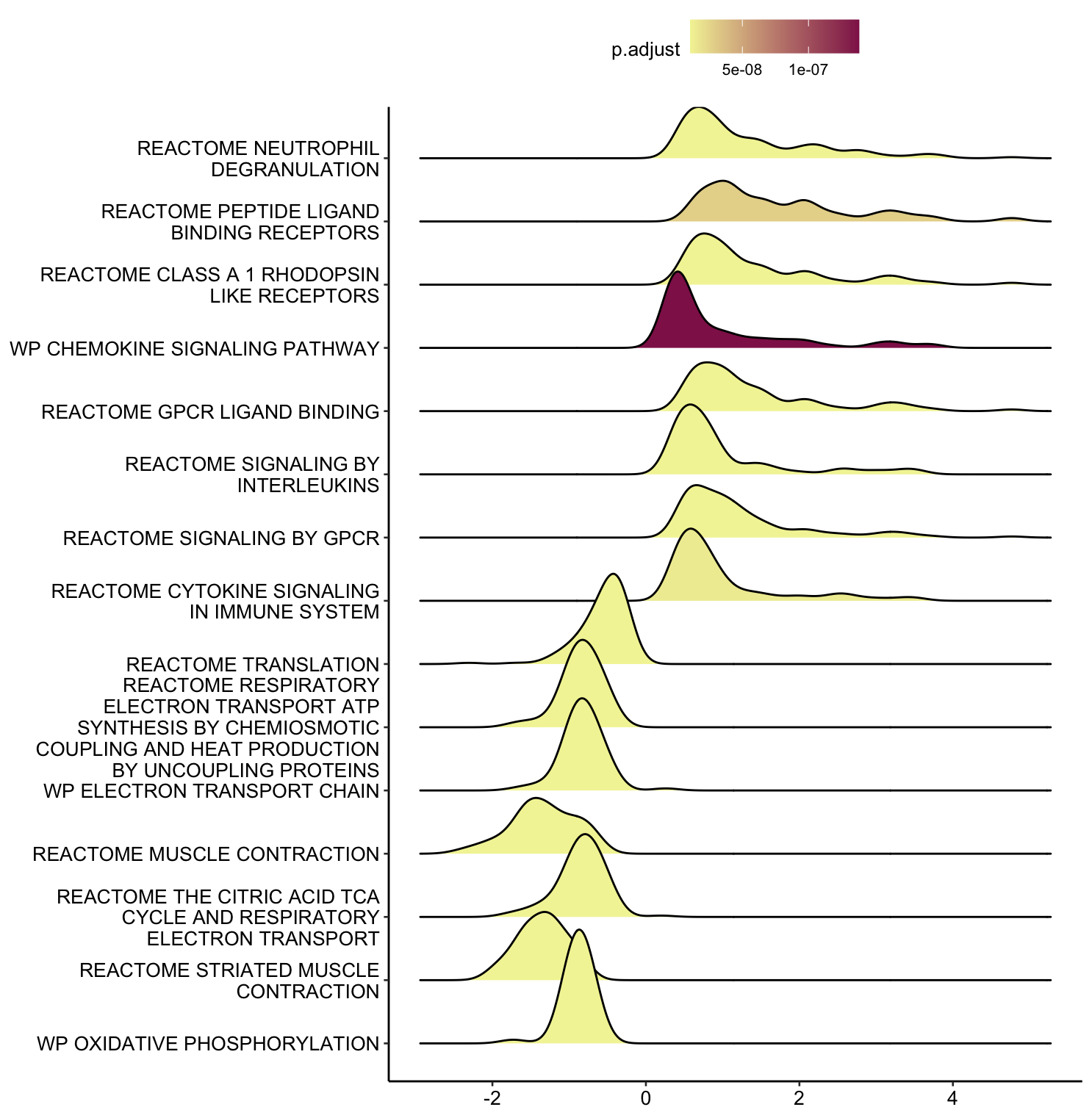
library(ggridges)
ridgeplot(gsea_results[[6]], showCategory=20)
## 更改字体大小和颜色
ridgeplot(gsea_results[[6]], showCategory=15)+scale_fill_gradient(low = "#F3F3A4", high = "#902058")
+theme_pubr(base_size = 10)
library(enrichplot) # https://mp.weixin.qq.com/s/HdY7yk4gOoofCFKY6DcdzA
gseaplot2(gsea_results[[11]], "GOBP_MEMBRANE_DEPOLARIZATION_DURING_CARDIAC_MUSCLE_CELL_ACTION_POTENTIAL")
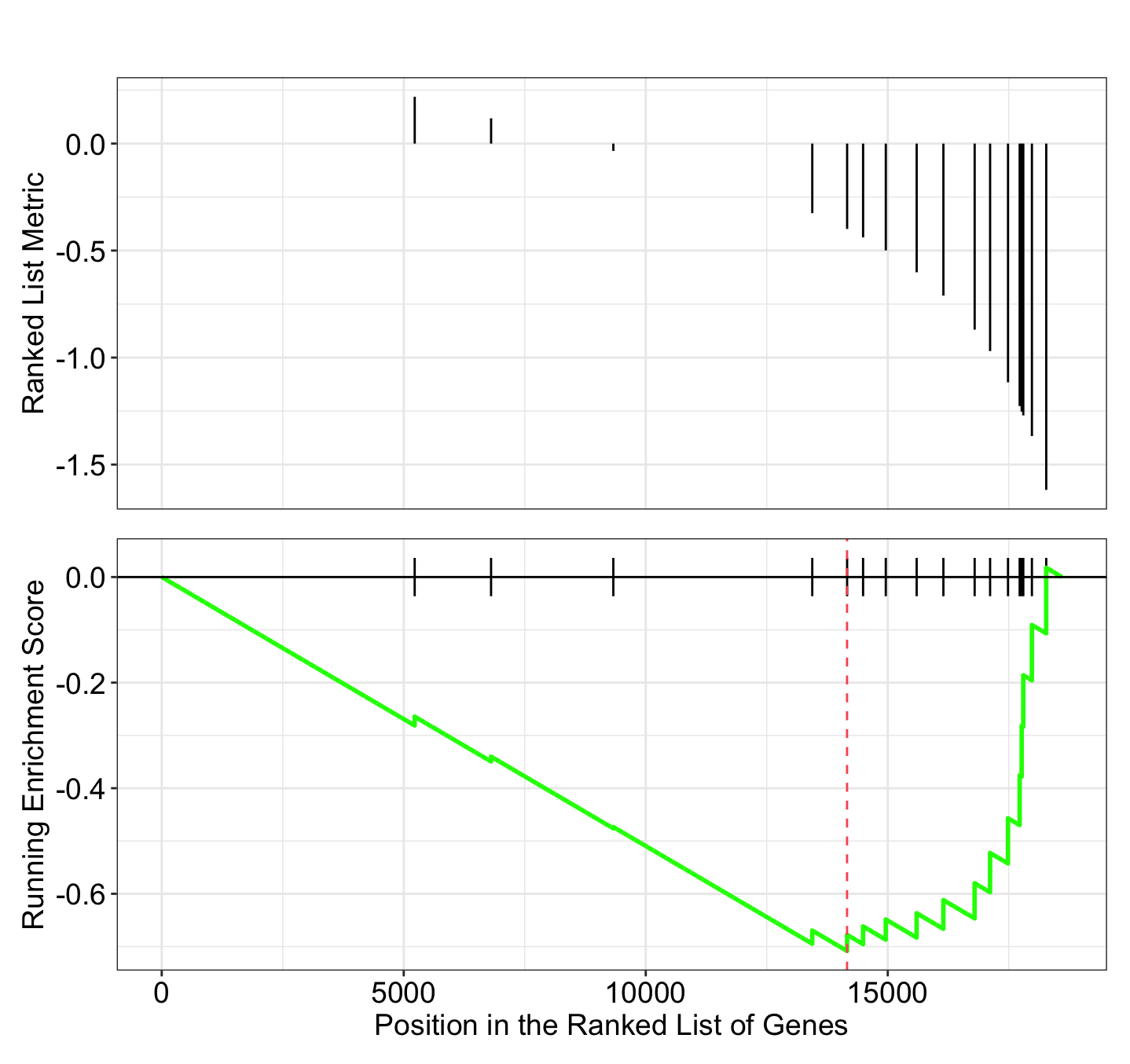
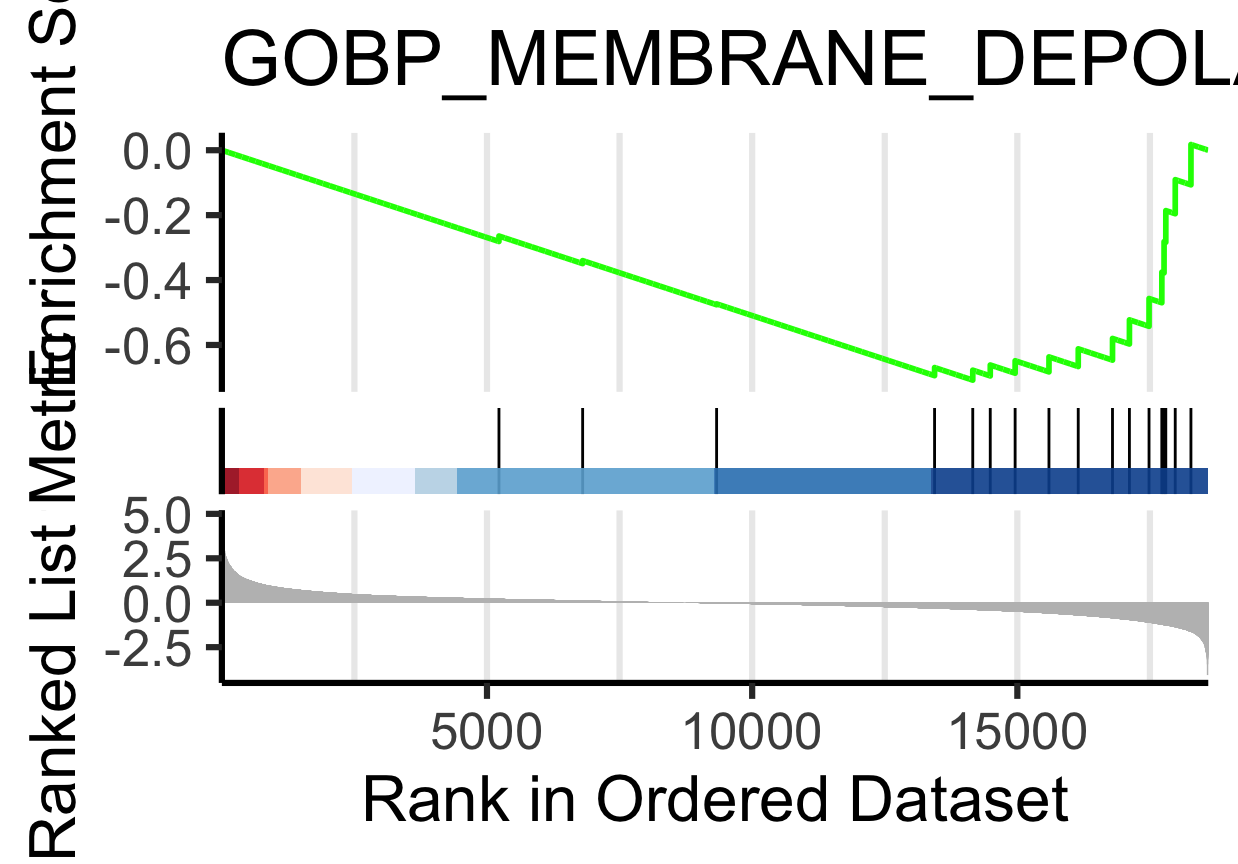
** (gseaplot) (gseaplot2)**
prerank <- read.table("result.txt",sep = "\t",header = T)
prerank$fcSign = sign(prerank$log2FoldChange)
prerank$logP = -log10(prerank$pvalue)
prerank$metric = prerank$logP/prerank$fcSign
y <- prerank[,c("genename", "metric")]
sum(is.na(y))
z = y[complete.cases(y),]
write.table(z,file="./Ydata/RNK.rnk",quote=F,sep="\t",row.names=F)
————————————————
版权声明:本文为CSDN博主「南柒北陆」的原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接及本声明。
原文链接:https://blog.csdn.net/ymy2019/article/details/105242040















 354
354











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









