








论文解读:《AFSE: towards improving model generalization of deep graph learning of ligand bioactivities targeting GPCR proteins》
文章地址:https://academic.oup.com/bib/article-abstract/23/3/bbac077/6554127?login=false
DOI:https://doi.org/10.1093/bib/bbac077
期刊:BRIEFINGS IN BIOINFORMATICS
影响因子/分区:9.5/二区
发布时间:2022年3月25日
Github:https://github.com/Yueming-Yin/AFSE
1.文章概述
配体分子自然地构成了图形结构。最近,许多优秀的深度图学习(deep graph learning,DGL)方法被提出并用于模拟配体生物活性,这对于从感兴趣的化合物数据库中虚拟筛选药物命中至关重要。然而,这些训练有素的 DGL 模型通常很难在真实场景中实现候选药物虚拟筛选的令人满意的性能。主要挑战包括训练模型的数据集规模小且有偏差,内部活跃的复杂案例会恶化模型性能。这些挑战将导致预测器过度拟合训练数据,并且在真实的虚拟筛选场景中泛化能力较差。因此,作者提出了一种名为对抗性特征子空间增强(adversarial feature subspace enhancement ,AFSE)的新算法。 AFSE通过双向对抗学习在新特征子空间中动态生成丰富的表示,然后最大程度地减少分子差异和生物活性的损失,以确保模型输出的局部平滑性,并显着增强DGL模型在预测配体生物活性方面的泛化能力。对七个最先进的开源 DGL 模型进行了基准测试,这些模型具有模拟配体生物活性的潜力,并通过多种标准进行精确评估。结果表明,在几乎所有 33 个 GPCR 数据集和 7 个 DGL 模型上,AFSE 极大地提高了其增强因子(top-10%、20% 和 30%),这是化合物数据库命中虚拟筛选中最重要的评估,同时保证了 RMSE 和 r2 上的优越性能。 AFSE 的 Web 服务器可在 http://noveldelta.com/AFSE 上免费获取。
2.关键点
- 在真实的虚拟筛选场景中,训练模型的数据集规模较小且有偏差,内部的活跃悬崖案例会恶化模型的泛化性能。
- AFSE通过双向对抗学习在新特征子空间中动态生成丰富的表示,然后最大限度地减少分子分歧和生物活性的最大损失,从而显着增强模型泛化能力。
- 在几乎所有33 G 蛋白偶联受体数据集和七个深度图学习模型上,AFSE 极大地提高了其增强因子、RMSE、r2 和τB。
- 在AFSE 中,所提出的处理两个子空间中每个特征的新机制已在理论和实践中得到证明,以确保模型输出函数更加泛化。
3.背景
药物分子可以自然地格式化为图形结构。与在 SMILES 格式上执行的典型基于序列的方法不同,基于图的算法倾向于挖掘隐藏在分子原子和键中的内部关系。
在这项研究中,作者重点关注以G蛋白偶联受体(GPCR)蛋白为靶点的配体分子,从 33 个人类代表性 GPCR 数据集中收集了配体,这些数据集涵盖了大多数人类 GPCR 家族。
为了说明 AFSE 的有效性,作者检查了所有七个具有预测配体生物活性能力的开源 DGL 模型的性能改进。从更多角度综合评估模型性能。评价指标包括常用来评价回归模型整体性能的均方根误差(RMSE)和相关系数(r2),以及药师在药物虚拟筛选中最关心的增强因子命中率(由于生物活性值的测试样本量较小,所以排在前10%、20%和30%),以及可以评估模型整体排名性能的Kendall tau-b系数。结果表明,在几乎所有 33 个 GPCRs 数据集和 7 个 DGL 模型上,AFSE 在保证 RMSE 和 r2 上的性能相当的情况下,极大地提高了模型的增强因子。
4.数据
所有活性和非活性配体数据集均从 GLASS 数据库下载。活性配体来源于“活性相互作用数据(IC50、EC50、Ki、Kd ≤ 10 uM)”文件,其中包括 674 个 GPCR 蛋白和独特的 616 545 个活性 GPCR 配体关联条目。
非活性配体来自“非活性相互作用数据(IC50、EC50、Ki、Kd > 10 uM)”文件,其中涉及 550 个 GPCR 蛋白和独特的 126 486 个非活性 GPCR 配体关联条目。
在本研究中使用定义为 − log10 v 的 p-生物活性值,其中 v 是原始生物活性(IC50、EC50、Ki、Kd)。无活性配体分子的生物活性值一般大于10 uM。
所有 743 031 个 GPCR 配体关联条目根据其 GPCR 蛋白和生物活性参数(IC50、EC50、Ki、Kd)分为 2802 个独立数据集,其中包含来自“活性相互作用数据”文件的 1659 个活性数据集和来自“非活性相互作用数据”文件的 1143 个非活性数据集。 作者在“非活动交互数据”文件里选择了 33 个有代表性的 GPCR 数据集,因为它们有足够的非活性配体。
作者根据以下两个原则构建训练和测试数据集: (i) 训练集(仅限活性):模拟用于训练的数据库,即大多数 GPCR 不包含足够的非活性配体分析,只分配训练集中的活性配体; (ii)测试集(#active<#inactive):为了模拟虚拟筛选的真实场景,即待筛选的数据库中往往含有大量的非活性配体。

5.方法
作者的 AFSE 方法由五个部分组成,涉及分子表示、生物活性预测、对抗性特征子空间增强、理论支持和算法实现(图 1)。原子和键的分子表示作为生物活性预测模型的初始输入;利用对抗性特征子空间增强来提高生物活性预测的模型泛化能力;理论支持是AFSE方法有效性的保证;算法实现是AFSE方法(算法1)的实现。
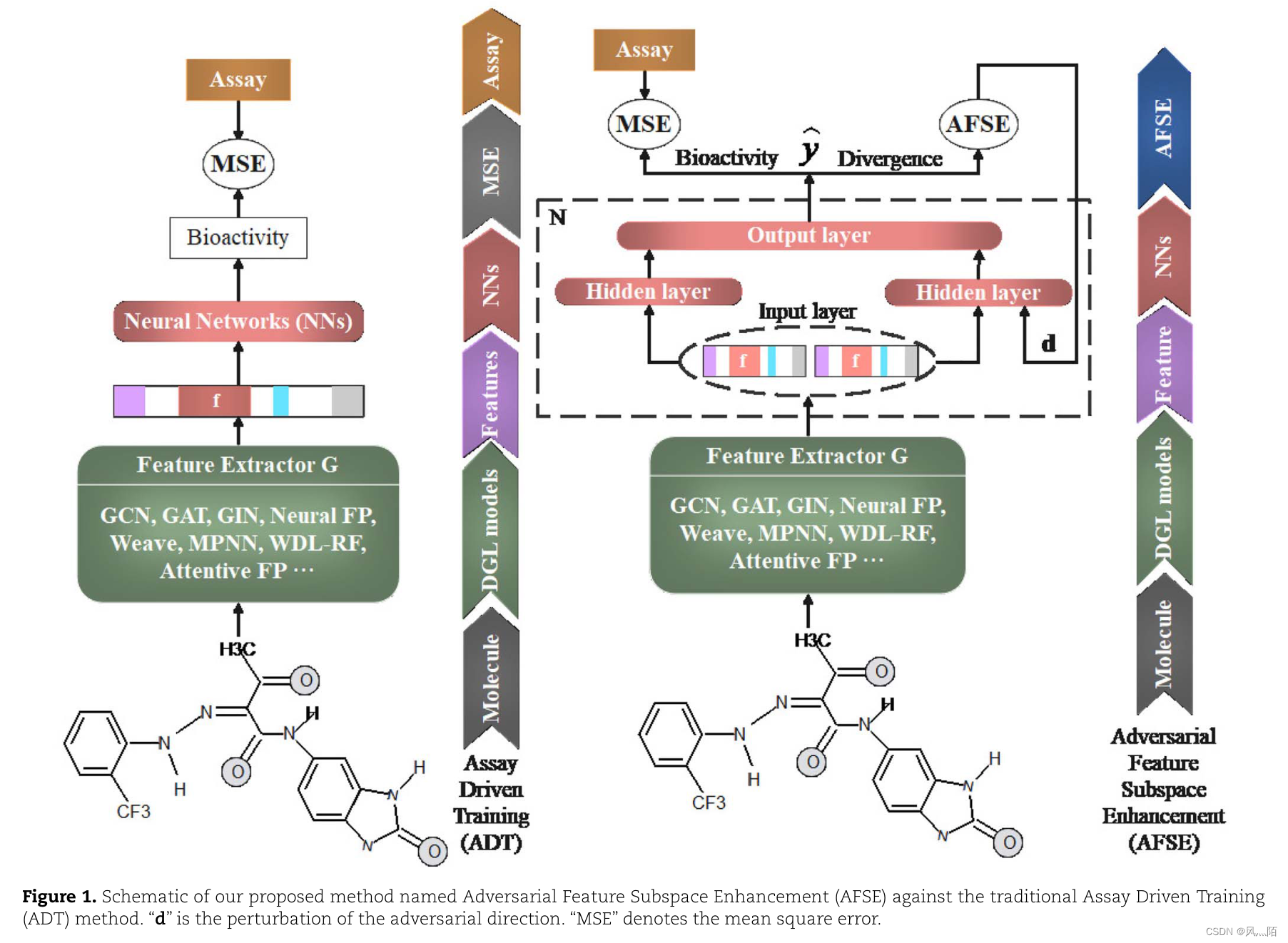
5.1 分子表示
在本研究中,使用规范的SMILES字符串来生成配体分子的拓扑结构,其由原子特征和键特征表示。
5.2 生物活性预测
作者提出了一种新颖的 AFSE 方法,该方法使深度图学习模型能够增强泛化能力,通过特征子空间中的对抗性训练来显着改进生物活性预测,并有效减轻对分析的依赖。该方法可以广泛部署到基于深度图学习的各种分子生物活性预测模型中,并显着提高预测精度和命中率。
在 AFSE 的两个子空间中处理每个特征,而不是仅在单个特征空间中。根据对抗训练理论,在训练过程中将随机扰动添加到输入中可以提高泛化性能。此外,在分析较少、缺乏监督信息的情况下,每个训练特征 f 的局部平滑度可以用来衡量泛化程度。两个特征子空间的散度小于仅一个特征空间的散度,这意味着在AFSE中,在两个子空间中处理每个特征的机制可以有效保证输出函数更加局部平滑。因此,AFSE 具有泛化能力,可以通过两个特征子空间中的对抗性训练来显着改进生物活性预测。
5.3 对抗性特征子空间增强
作者定义一个散度函数来评估每个特征的局部平滑度,特别是对于没有 Assay 的特征。
将对抗方向d的扰动输入到每个特征f中,以尽可能增加输出函数的散度,同时深度图网络G和神经网络N通过反向传播来减少整个损失函数。通过这样的对抗性训练,输出函数变得更加平滑,AFSE变得不易受到小偏差的影响,这可以显着提高泛化性能。
5.4 算法实现
神经网络N由输入层、隐藏层和输出层组成。如果将深度图网络 G 提取的特征输入到 N 中,则可以将预测的生物活性 ^y 计算为 N 的输出。特征 f 被投影到两个不同的子空间中,然后将两个投影向量相加。
5.5 评价指标
增强因子(Enhancement factor,EF)是药剂师在药物命中虚拟筛选中最关心的指标。
均方根误差 (The root mean square error,RMSE) 指标是评估回归模型的通用指标,RMSE 越低,模型预测的平均准确度越高。
采用相关系数(r2)指标来评估 Kaggle 2012 期间参与者的活动预测表现,r2越大,模型预测的整体精度越高。
6.结果
6.1 实验设置
作者选择了七个最先进的 DGL 模型作为G,包括图卷积网络(GCN)、图注意网络(GAT)、图同构网络(GIN)、通过监督学习和上下文预测进行预训练、神经指纹(Neural FP)、Weave、消息传递神经网络(MPNN)和注意力FP。这些 DGL 模型的开源代码和用法可以在 Github上找到。为了公平比较, AFSE 方法和所有 DGL 模型上的 ADT 方法都分配了相同的参数设置。
6.2 小规模和有偏差数据集的比较
为了缓解训练样本的分布偏差,可以在表1中观察到训练数据集和测试数据集的生物活性分布之间的差异,这进一步揭示了训练样本分布偏差的问题。以小规模数据集的Task 7为例,其中的生物活性范围之间存在明显的偏差。

作者还提供了分别采用传统 ADT 和提出的 AFSE 时特征分布的可视化。如图3所示,当采用ADT时,样本分散在整个特征空间上,并且训练和测试数据集的特征分布无法相互对齐,这验证了ADT没有充分解决有偏差的问题。相反,AFSE可以通过在两个特征子空间中进行对抗性训练来确保特征空间更加局部平滑,如图3所示,这可以保证AFSE更好的泛化能力,从而有效解决有偏差的问题。与ADT相比,特征中心之间的间隔减少了26.2%。

6.3 增强因子与τB的比较

为了直观地呈现模型性能,图 4 显示了使用 AFSE 的 DGL 模型与使用 ADT 的模型在所有数据集上的 EF 指标的平均改进。结果表明,所有采用 AFSE 的 DGL 模型的 EF 指标均高于采用 ADT 的模型,这表明 AFSE 通常可以增强 DGL 模型,以虚拟筛选更有前景的候选药物。

为了系统地验证我们的 AFSE 方法击中高活性化合物的能力,我们在表 3 中总结了 AFSE 在不同大小和偏差的数据集上实现的 EF10% 和 τB 指标的改进,其中 ADT 的性能用作基线,并对七个网络的性能进行平均。结果表明,AFSE 在不同规模和偏差程度的数据集上带来了性能提升。其中,小样本数据集上的改进最为显着。
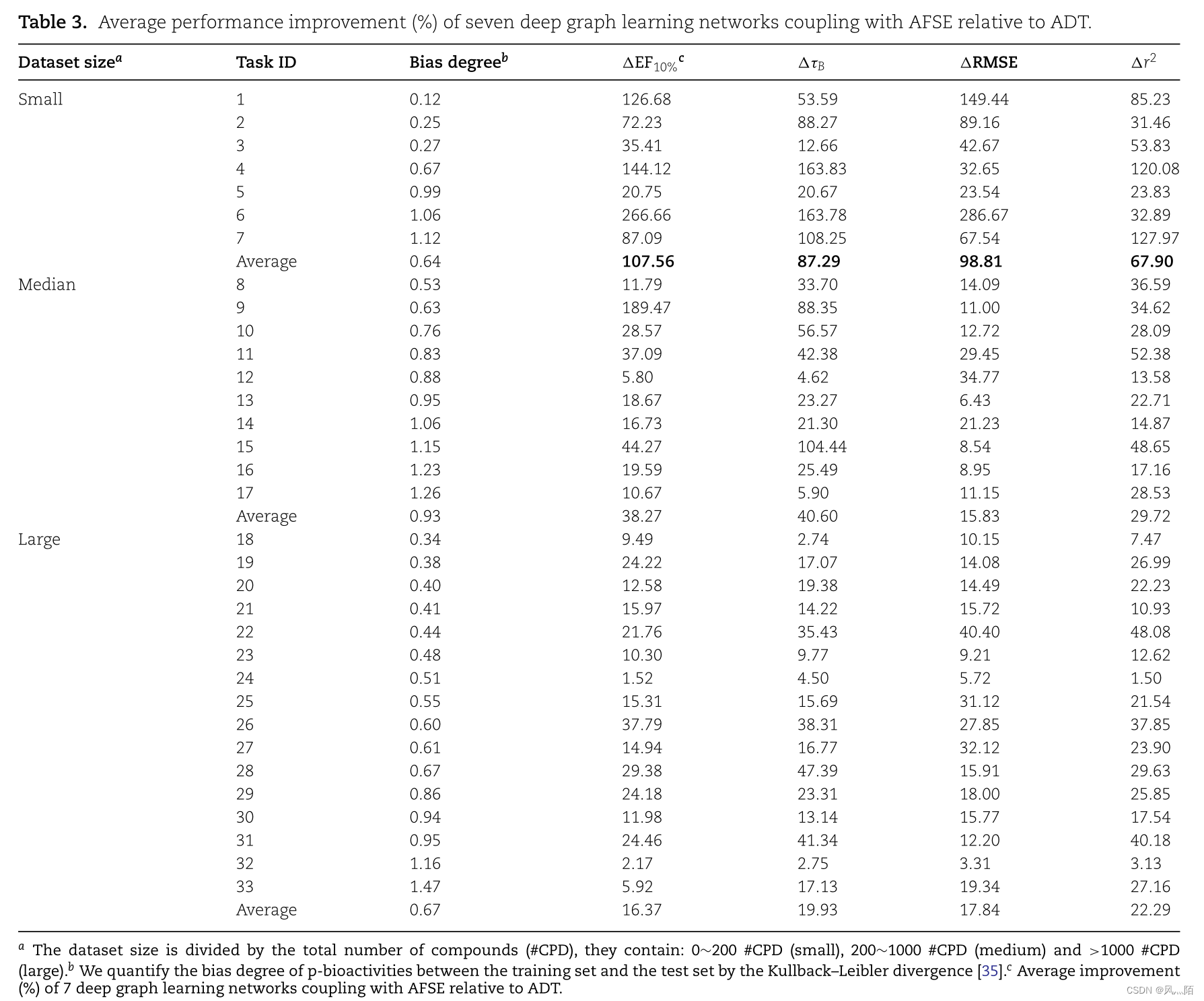
6.4 RMSE 和 r2 的比较
为了系统地验证我们的 AFSE 方法准确预测化合物生物活性的能力,我们还在表 3 中总结了 AFSE 在不同大小和偏差的数据集上的 RMSE 和 r2 改进。
6.5 训练过程可视化

6.6 虚拟筛选的实际应用
以 GAT 为例,图 6 显示了通过利用 Drugbank 数据库和 AFSE 方法的自定义数据库,各种数据集在重要 EF10% 和 r2 指标上的性能改进。三种基于 AFSE 的 DGL 模型(AFSE、AFSE by Drugbank 数据库和 AFSE by Custom Database)具有不同的性能,但与基于 ADT 的 DGL 模型相比,它们都表现出优越的性能来验证 AFSE 的泛化能力。

7.结论
作者开发了一种名为 AFSE 的新颖算法,它可以通过双向对抗性学习动态学习特征向量的充分表示,并最大限度地减少分子分歧和生物活性的最大损失。
在七个优秀的开源 DGL 模型上测试了大型基准,这些模型具有预测配体生物活性的能力。
在几乎所有 33 个 GPCR 数据集和 7 个 DGL 模型上,AFSE 显着提高了其增强因子(前 10%、20% 和 30%),同时实现了更好的效果RMSE 和 r2 的性能。
未来发展方向:
- 3D-QSAR技术,可以将化合物分子的空间信息添加到DGL模型中,如原子的三维坐标信息、取向、扭转、二面角等。
- 自由能扰动将引入技术来分析当与一种化合物中原子或原子团的微小变化耦合时配体-蛋白质相互作用的自由能变化,并进一步获得其对配体-蛋白质结合亲和力的影响














 7525
7525











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









