







网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
- 安装编译所需的依赖项:
sudo apt install build-essential cmake git
- 克隆 NGLess 仓库:
git clone https://github.com/ngless-toolkit/ngless.git
- 进入目录并编译安装:
cd ngless
make
sudo make install
方法三:conda环境安装
conda环境安装可参考:轻快小miniconda3在linux下的安装配置-centos9stream-Miniconda3 Linux 64-bit-CSDN博客
conda create -n ngless_env python=3
conda activate ngless
conda install -c bioconda ngless
# 或mamba
mamba create -n ngless_env python=3
mamba activate negless
mamba install -c bioconda ngless
方法四:docker安装
docker pull ngless/ngless
docker run -it ngless/ngless
注意事项:
- 在 Docker 中运行 NGLess,您可以在容器中执行各种 NGLess 操作,但默认情况下,容器中的任何更改都不会保留。如果希望保留结果或输出文件,请将宿主机的文件夹与 Docker 容器中的文件夹进行挂载。
- 若要将宿主机的文件夹挂载到 Docker 容器中,可以使用
-v参数。例如:
docker run -it -v /path/to/host/folder:/path/to/container/folder ngless/ngless
这将把宿主机的 /path/to/host/folder 目录挂载到容器的 /path/to/container/folder 目录中。
在 macOS 上安装 NGLess:
在 macOS 上安装 NGLess 可以使用 Homebrew 或手动编译安装:
方法一:使用 Homebrew 安装
- 安装 Homebrew(如果尚未安装):
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"
- 使用 Homebrew 安装 NGLess:
brew install ngless
方法二:从源代码编译安装
- 安装 Xcode Command Line Tools:
xcode-select --install
- 安装编译所需的依赖项(使用 Homebrew):
brew install cmake
- 克隆 NGLess 仓库并编译安装:
git clone https://github.com/ngless-toolkit/ngless.git
cd ngless
make
sudo make install
在 Windows 上安装 NGLess:
在 Windows 上可以通过虚拟机、Docker 或 Windows Subsystem for Linux(WSL)来运行 NGLess。以下是使用 WSL 安装 NGLess 的步骤:
- 启用 WSL 和安装 Linux 发行版(如 Ubuntu):请参考 Microsoft 官方文档中关于 WSL 的说明和安装步骤。
- 安装 NGLess(可参考前述 Linux 安装方法):在 WSL 中按照 Linux 的安装步骤进行操作。
NGLess常用功能函数
NGLess Builtin Functions — NGLess 1.5.0 documentation
NGLess是一个用于处理和分析NGS(Next-Generation Sequencing)数据的编程语言和工具。下面是NGLess的一些常用函数和示例代码:
count() - 计算一个序列文件中的序列数量
input = fastq('input.fastq.gz')
reads = count(input)
write() - 将结果写入到一个输出文件
input = fastq('input.fastq.gz')
write(input, ofile='output.fastq')
parse_fastq() - 解析FASTQ文件
input = parse_fastq('input.fastq')
select() - 选择符合条件的序列
input = fastq('input.fastq.gz')
selected = select(input, keep_if=(length >= 50))
reject() - 拒绝符合条件的序列
input = fastq('input.fastq.gz')
filtered = reject(input, keep_if=(mean_quality < 20))
substrim() - 对序列进行质量截断
input = fastq('input.fastq.gz')
trimmed = substrim(input, cutoff=20)
map() - 对序列进行比对
input = fastq('input.fastq.gz')
index = faidx('reference.fasta')
mapped = map(input, index)
unmapped_only() - 选择未比对的序列
input = fastq('input.fastq.gz')
mapped = map(input, index)
unmapped = unmapped_only(mapped)
group_by() - 按照指定的键值进行分组
input = ... # 一些数据
grouped = group_by(input, key='sample')
sort() - 对序列进行排序
input = ... # 一些数据
sorted_data = sort(input, by='value', reverse=True)
mean() - 计算一组数字的平均值
data = [1, 2, 3, 4, 5]
avg = mean(data)
median() - 计算一组数字的中位数
data = [1, 2, 3, 4, 5]
med = median(data)
sum() - 计算一组数字的总和
data = [1, 2, 3, 4, 5]
total = sum(data)
range() - 生成一个整数序列
numbers = range(1, 10)
length() - 计算序列或字符串的长度
seq = 'ATCG'
len = length(seq)
reverse_complement() - 对序列取反补
seq = 'ATCG'
rc_seq = reverse_complement(seq)
translate() - 将DNA序列翻译成氨基酸序列
dna_seq = 'ATGCTGAACTG'
aa_seq = translate(dna_seq)
gc_content() - 计算序列的GC含量
seq = 'ATCGATCG'
gc = gc_content(seq)
subsample() - 对序列进行子抽样
input = fastq('input.fastq.gz')
subsampled = subsample(input, fraction=0.1)
merge() - 合并两个或多个序列文件
input1 = fastq('input1.fastq.gz')
input2 = fastq('input2.fastq.gz')
merged = merge(input1, input2)
average_quality() - 计算序列的平均质量
input = fastq('input.fastq.gz')
avg_qual = average_quality(input)
trim_polya() - 对序列进行poly(A)尾修剪
input = fastq('input.fastq.gz')
trimmed = trim_polya(input)
annotate() - 对序列进行注释
input = ... # 一些序列
annotation = ... # 一些注释信息
annotated = annotate(input, with=annotation)
to_fasta() - 将序列文件转换为FASTA格式
input = fastq('input.fastq.gz')
fasta = to_fasta(input)
to_fastq() - 将序列文件转换为FASTQ格式
input = fasta('input.fasta')
fastq = to_fastq(input)
reverse() - 对序列进行反转
seq = 'ATCG'
reversed_seq = reverse(seq)
complement() - 对序列进行互补
seq = 'ATCG'
comp_seq = complement(seq)
is_paired() - 判断序列是否成对出现
input = fastq('input.fastq.gz')
paired = is_paired(input)
pair() - 对成对的序列进行配对
input = fastq('input.fastq.gz')
paired = pair(input)
is_unique() - 判断序列是否唯一
input = fastq('input.fastq.gz')
unique = is_unique(input)
unique_only() - 选择唯一的序列
input = fastq('input.fastq.gz')
unique = unique_only(input)
random() - 生成一个随机数
rand_num = random()
shuffle() - 对序列进行随机重排
input = fastq('input.fastq.gz')
shuffled = shuffle(input)
annotate_gff() - 对序列进行基因组注释
input = fasta('input.fasta')
gff_file = 'annotation.gff'
annotated = annotate_gff(input, gff_file)
align() - 对序列进行局部或全局比对
input = fasta('input.fasta')
ref_seq = fasta('reference.fasta')
alignment = align(input, ref_seq)
align_sam() - 对序列进行SAM格式比对
input = fasta('input.fasta')
sam_file = 'alignment.sam'
aligned = align_sam(input, sam_file)
align_bam() - 对序列进行BAM格式比对
input = fasta('input.fasta')
bam_file = 'alignment.bam'
aligned = align_bam(input, bam_file)
compress() - 对文件进行压缩
input_file = 'input.txt'
compressed_file = compress(input_file, format='gzip')
decompress() - 对文件进行解压缩
compressed_file = 'input.gz'
decompressed_file = decompress(compressed_file)
distance() - 计算两个序列之间的距离
seq1 = 'ATCG'
seq2 = 'AGCG'
dist = distance(seq1, seq2)
intersect() - 计算两个序列集合的交集
input1 = fasta('input1.fasta')
input2 = fasta('input2.fasta')
intersected = intersect(input1, input2)
union() - 计算两个序列集合的并集
input1 = fasta('input1.fasta')
input2 = fasta('input2.fasta')
unioned = union(input1, input2)
subtract() - 计算两个序列集合的差集
input1 = fasta('input1.fasta')
input2 = fasta('input2.fasta')
subtracted = subtract(input1, input2)
average() - 计算一组数字的平均值
data = [1, 2, 3, 4, 5]
avg = average(data)
maximum() - 计算一组数字的最大值
data = [1, 2, 3, 4, 5]
max_val = maximum(data)
minimum() - 计算一组数字的最小值
data = [1, 2, 3, 4, 5]
min_val = minimum(data)
standard_deviation() - 计算一组数字的标准差
data
常见生信分析代码片段
下面是常用的NGless功能函数处理宏基因组数据的代码片段:
从FASTQ文件中过滤低质量的reads,并将结果输出到新文件中:
ngless "1.0"
input = fastq('input.fq')
preprocess(input, phred=33) using |read|:
if read.avg_qual < 20:
discard
input | keep | add_sequence_length | sum | write(`filtered.fq`, compression=Fastq)
根据OTU表将reads映射到参考数据库,并生成OTU表:
ngless "1.0"
input = fastq('input.fq')
reference = fasta('ref.fasta')
mapped = map(input, reference, exact=False, sensitive=True)
otu_table(mapped, reference) | write(`otu_table.txt`, format="csv")
对OTU表进行物种注释,并生成注释表:
ngless "1.0"
otu_table = csv('otu_table.csv')
annotation_db = csv('annotation_db.csv')
annotated = annotate_species(otu_table, annotation_db)
annotated | write(`annotated_otu_table.csv`, format="csv")
根据OTU丰度信息生成热图:
ngless "1.0"
otu_table = csv('otu_table.csv')
heatmap(otu_table) | write(`heatmap.png`, format="png")
对样品进行稀释,并生成稀释后的OTU表:
ngless "1.0"
otu_table = csv('otu_table.csv')
diluted = dilute(otu_table, factor=10)
diluted | write(`diluted_otu_table.csv`, format="csv")
对OTU表进行组间差异分析,使用差异显著性检验方法:
ngless "1.0"
otu_table = csv('otu_table.csv')
groups = csv('groups.csv')
differential(abundance(otu_table), groups) | write(`differential_analysis.csv`, format="csv")
对样品进行Beta多样性分析,并生成PCoA图:
ngless "1.0"
otu_table = csv('otu_table.csv')
pcoa_table = pcoa(otu_table)
pcoa_table | plot(`pcoa.png`, format="png")
对OTU表进行功能注释,并生成功能注释表:
ngless "1.0"
otu_table = csv('otu_table.csv')
gene_db = csv('gene_db.csv')
annotated = annotate_functions(otu_table, gene_db)
annotated | write(`annotated_otu_table.csv`, format="csv")
根据OTU表进行物种多样性分析,并生成物种多样性指数表:
ngless "1.0"
otu_table = csv('otu_table.csv')
diversity_indices(otu_table) | write(`diversity_indices.csv`, format="csv")
对样品进行Alpha多样性分析,并生成稀释曲线图:
ngless "1.0"
otu_table = csv('otu_table.csv')
alpha_table = alpha_diversity(otu_table)
alpha_table | plot(`alpha_diversity.png`, format="png")
对OTU表进行物种丰度分析,并生成物种丰度柱状图:
ngless "1.0"
otu_table = csv('otu_table.csv')
species_abundance(otu_table) | plot(`species_abundance.png`, format="png")
根据OTU表进行共生网络分析,并生成共生网络图:
ngless "1.0"
otu_table = csv('otu_table.csv')
cooccurrence_network(otu_table) | plot(`cooccurrence_network.png`, format="png")
对样品进行微生物组成分析,并生成样品组成饼图:
ngless "1.0"
otu_table = csv('otu_table.csv')
sample_composition(otu_table) | plot(`sample_composition.png`, format="png")
对OTU表进行代谢通路分析,并生成代谢通路富集柱状图:
ngless "1.0"
otu_table = csv('otu_table.csv')
pathway_db = csv('pathway_db.csv')
enriched_pathways(otu_table, pathway_db) | plot(`enriched_pathways.png`, format="png")
对OTU表进行进化分析,并生成进化树:
ngless "1.0"
otu_table = csv('otu_table.csv')
evolutionary_analysis(otu_table) | plot(`evolutionary_tree.png`, format="png")
将多个OTU表进行合并:
ngless "1.0"
otu_table1 = csv('otu_table1.csv')
otu_table2 = csv('otu_table2.csv')
combined = merge(otu_table1, otu_table2)
combined | write(`merged_otu_table.csv`, format="csv")
对OTU表进行样品分类,并生成分类树:
ngless "1.0"
otu_table = csv('otu_table.csv')
taxonomy_tree(otu_table) | plot(`taxonomy_tree.png`, format="png")
根据OTU表进行功能富集分析,并生成功能富集柱状图:
ngless "1.0"
otu_table = csv('otu_table.csv')
gene_set_db = csv('gene_set_db.csv')
enriched_functions(otu_table, gene_set_db) | plot(`enriched_functions.png`, format="png")
对样品进行Gamma多样性分析,并生成Gamma多样性曲线:
ngless "1.0"
otu_table = csv('otu_table.csv')
gamma_table = gamma_diversity(otu_table)
gamma_table | plot(`gamma_diversity.png`, format="png")
对OTU表进行进化树构建,并生成进化树:
ngless "1.0"
otu_table = csv('otu_table.csv')
phylogenetic_tree(otu_table) | plot(`phylogenetic_tree.png`, format="png")
请注意,这些代码片段仅为示例,并不一定适用于所有NGless版本和数据集。在实际使用时,请根据具体情况进行修改和调整。
NGLess的使用示例
人类肠道宏基因组学的功能和分类分析
步骤概述:
- 准备数据:获取并准备用于分析的原始测序数据。
- 质量控制和过滤:对原始数据进行质量控制、去除低质量序列和去除宿主DNA等。
- 人类肠道宏基因组分类:对数据进行分类,识别宿主肠道微生物。
- 功能注释:对宏基因组数据进行功能注释,识别和分析肠道微生物的功能特征。
详细步骤:
步骤 1: 准备数据
- 下载并准备用于肠道宏基因组学的原始测序数据,例如来自人类肠道样本的元组装数据(metagenomic sequencing data)。
步骤 2: 质量控制和过滤
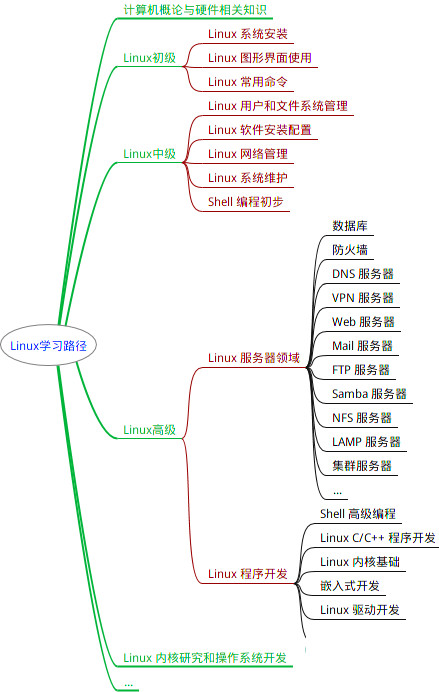
最全的Linux教程,Linux从入门到精通
======================
-
linux从入门到精通(第2版)
-
Linux系统移植
-
Linux驱动开发入门与实战
-
LINUX 系统移植 第2版
-
Linux开源网络全栈详解 从DPDK到OpenFlow

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!















 5万+
5万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









