






1. 什么是热图?
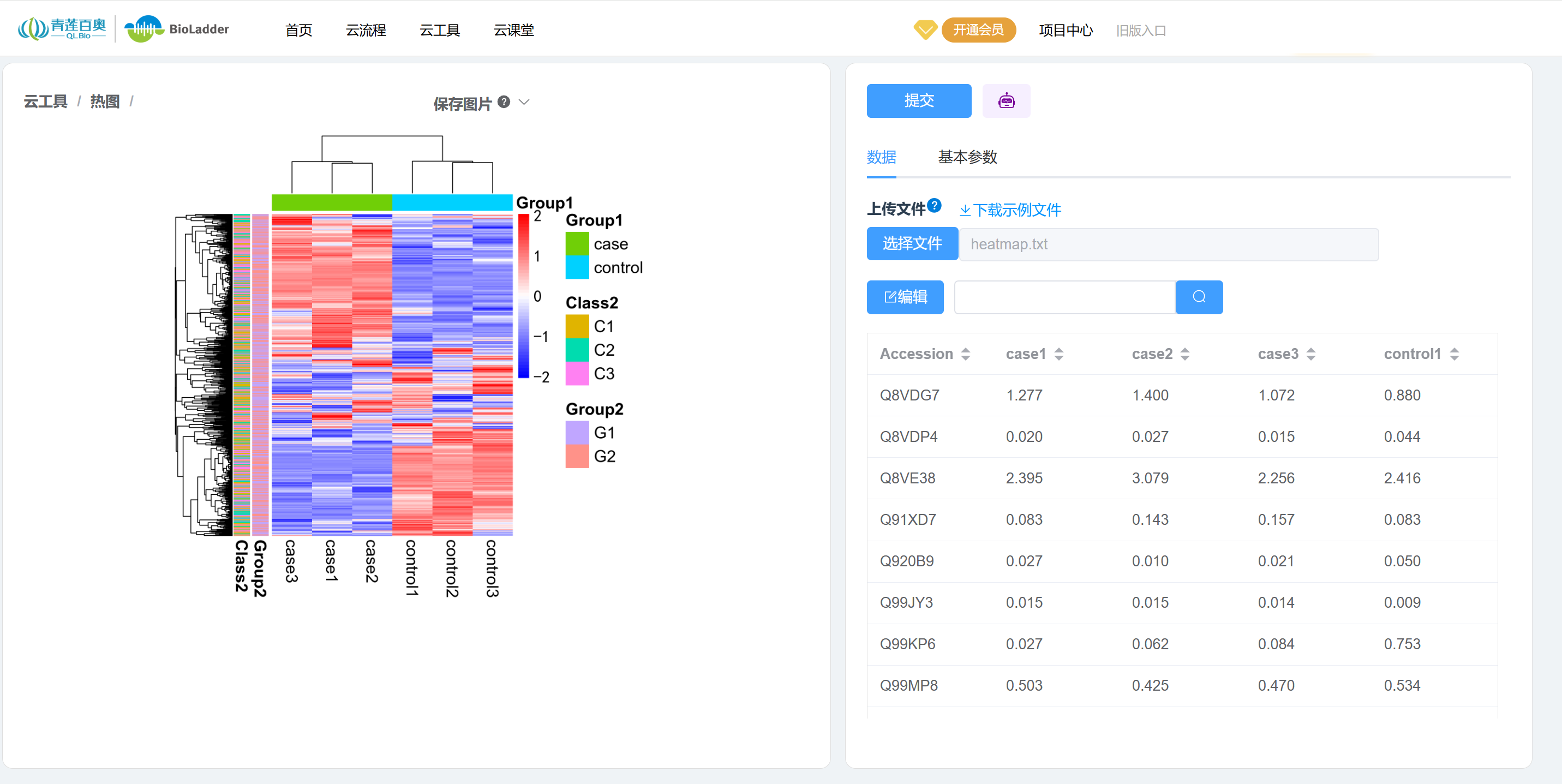
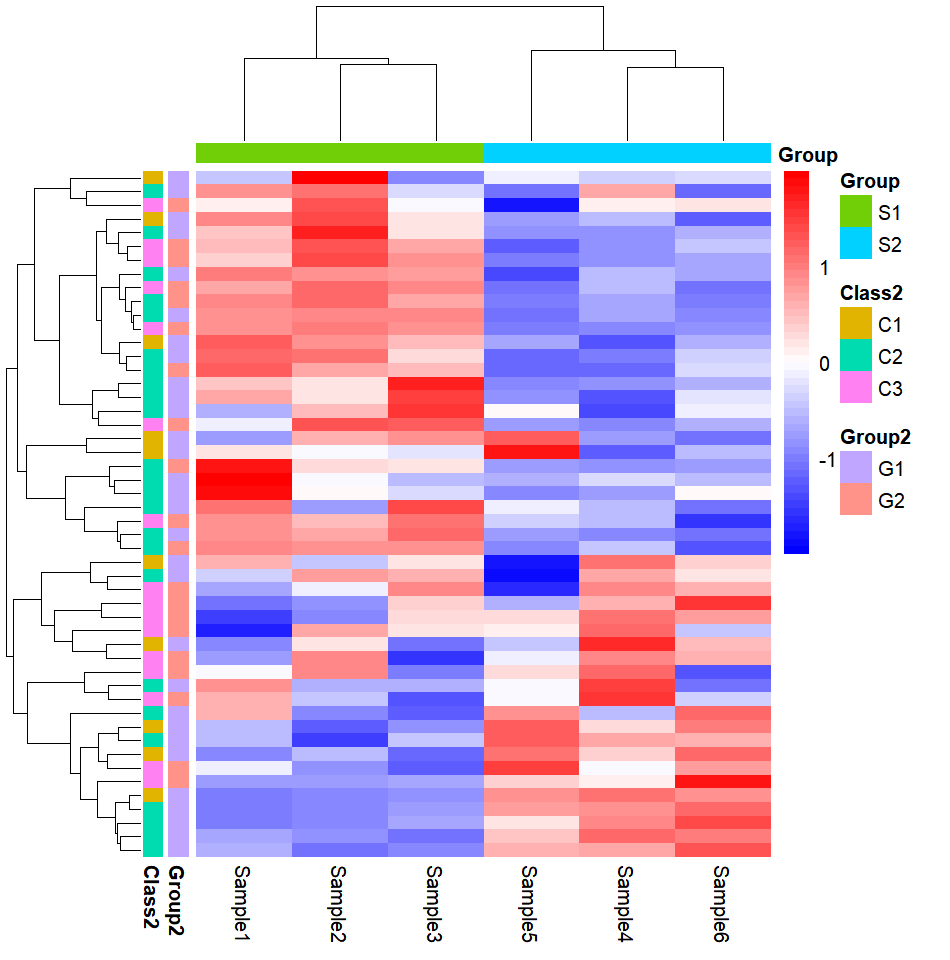
如图,就是一副组学研究中热图的常用绘制模式,每个小方格表示每个基因在不同样本中的定量值,其颜色表示该基因表达量大小,红色为高表达,蓝色为低表达。

- 行名称:通常为样本名称。
- 列名称:通常为基因名称。
- 图例信息:左侧图例表示热图表达量的颜色说明,右侧图例展示分组信息。热图绘制过程中通常会进行Z-score归一化处理,因此图例数据显示为0左右对称分布,这表示已进行Z-score标准化。
- 列聚类:如果不进行聚类,列的排序将保持原始数据的顺序。通过聚类可以判断不同组别的样本是否被正确分组。理论上,来自同一组的样本应该表现出相似的特征。如果某样本被错误地聚类到其他组,可能意味着该样本在变异度上较大,或者在样本采集或测序等过程中存在问题。
- 行聚类:如果不进行聚类,行的排序将保持原始数据的顺序。聚类分析可以帮助识别具有一致表达模式的基因群体。
- 列分组信息:展示样本的分组标签,通常用于标明不同条件或类别的样本。
- 行分组信息:展示基因的分组信息,通常用于表示基因的功能类别或其他分类信息。
2. 绘图的数据准备
分为绘图数据与分组数据,其中分组数据为非必须的。
示例数据可以在https://www.bioladder.cn/web/heatmap.txt找到并下载。
2.1. 热图数据
数据来源一般是搜库结果定量表。包含2个维度的数据,一般情况下,每一行是一个基因,每一列是一个样本。
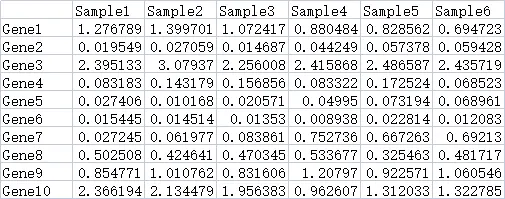
2.2. 样本分组数据(可选)
行名的名称和个数要和之前的heatmap数据保持一致,列名为分组名称,可以包含不止一个分组。
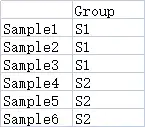
2.3. 基因分组数据(可选)
行名的名称和个数要和之前的heatmap数据保持一致,列名为分组名称,可以包含不止一个分组。
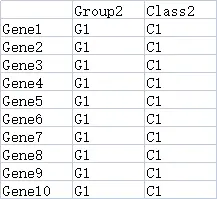
3. 绘制热图的方法
3.1. BioLadder v2.0 云平台在线绘图
BioLadder v2.0 是一个在线绘图平台,我们只需要上传数据,系统自动生成图表,不需要写代码,适合0代码基础的同学。网址:BioLadder v2.0-生物信息在线分析云平台
3.2. R语言绘制热图
喜欢自己写代码的同学,也附上R语言代码可供参考。
library(pheatmap) # 加载pheatmap这个R包
# 1,读取热图数据文件
df = read.delim("https://www.r2omics.cn/res/demodata/heatmap/data.heatmap.txt", #文件名称 注意文件路径,格式
header = T, # 是否有标题
sep = "\t", # 分隔符是Tab键
row.names = 1, # 指定第一列是行名
fill=T) # 是否自动填充,一般选择是
# (可选)读取分组数据文件
dfSample = read.delim("https://www.r2omics.cn/res/demodata/heatmap/sample.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
dfGene = read.delim("https://www.r2omics.cn/res/demodata/heatmap/gene.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
# 2,绘图
pheatmap(df,
annotation_row=dfGene, # (可选)指定行分组文件
annotation_col=dfSample, # (可选)指定列分组文件
show_colnames = TRUE, # 是否显示列名
show_rownames=TRUE, # 是否显示行名
fontsize=5, # 字体大小
color = colorRampPalette(c('#0000ff','#ffffff','#ff0000'))(50), # 指定热图的颜色
annotation_legend=TRUE, # 是否显示图例
border_color=NA, # 边框颜色 NA表示没有
scale="row", # 指定归一化的方式。"row"按行归一化,"column"按列归一化,"none"不处理
cluster_rows = TRUE, # 是否对行聚类
cluster_cols = TRUE # 是否对列聚类
)
这段代码通过pheatmap包绘制热图,主要步骤是:
- 使用read.delim()加载热图数据和分组信息(可选的行列分组)。
- 使用pheatmap()绘制热图,设置颜色、显示行列名、归一化方式(按行或按列)以及是否进行聚类等参数。
最终生成带有分组信息和聚类的热图。

















 902
902

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









