







欢迎大家关注全网生信学习者系列:
- WX公zhong号:生信学习者
- Xiao hong书:生信学习者
- 知hu:生信学习者
- CDSN:生信学习者2
介绍
中介分析(Mediation Analysis)是一种统计方法,用于研究一个自变量(通常是独立变量或预测变量)如何通过一个或多个中介变量(也称为中介因素或中介机制)影响因变量(通常是响应变量或结果变量)。中介分析的目的是揭示变量之间的内在关系,特别是自变量对因变量的间接效应,以及这种效应是如何通过中介变量传递的。
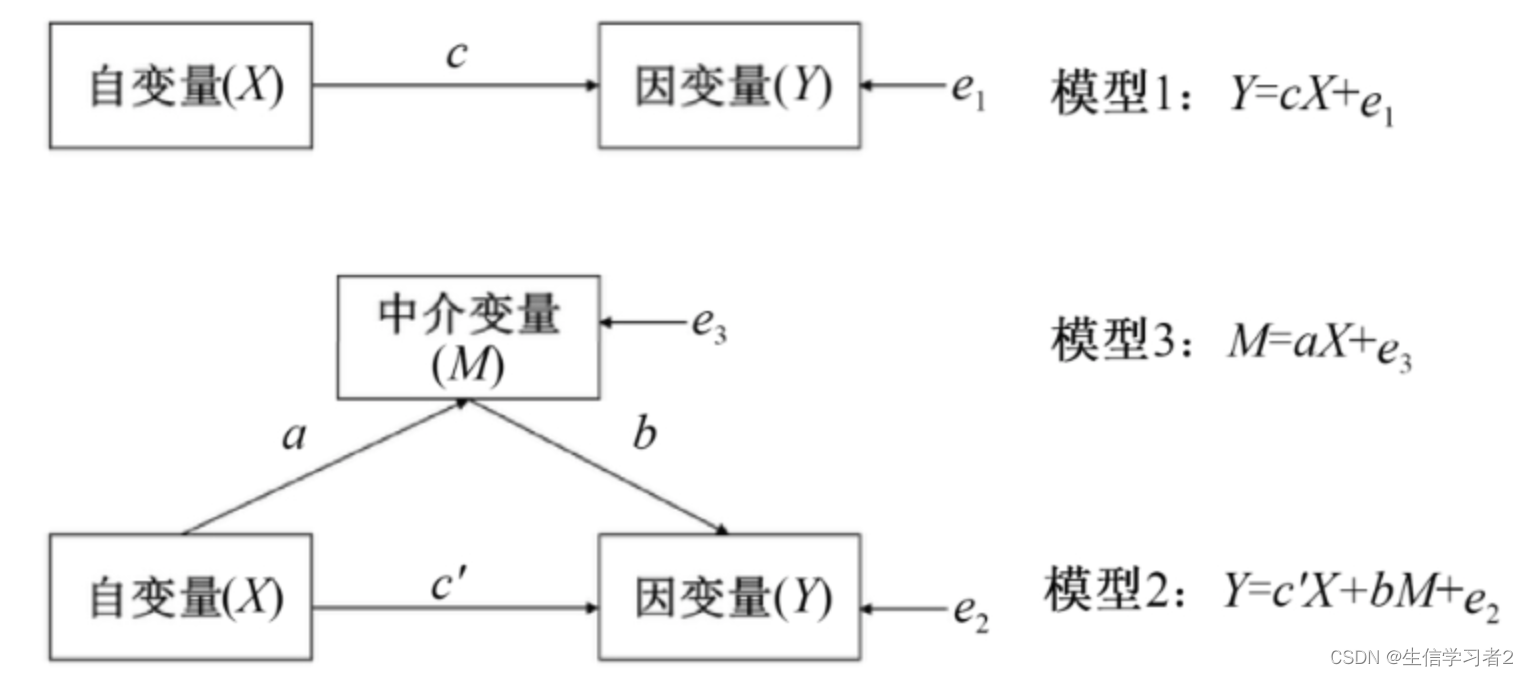
评估识别出的与结局变量显著相关的标记物如炎症细胞因子cytokines、肠道微生物gut microbiota和短链脂肪酸SCFA是否能够在伴侣数目number of partners和HIV-1血清转化HIV-1 seroconversion之间起到中介作用。
自然效应模型(Natural Effect Model)是一种统计方法,用于估计在自然情况下(即在没有干预或随机分配的情况下)变量之间的因果关系。在流行病学和临床研究中,这种模型特别有用,因为它可以帮助研究者了解不同因素对健康结果的自然影响。以下是中介分析的变量解析:
-
exposure variables (自变量X): consisting of sexual exposure groups
-
mediators (中介变量M): biomarkers (cytokines, gut microbiota, SCFA)
-
outcome variable (因变量Y): HIV-1 seroconversion status
加载R包
library(readr)
library(openxlsx)
library(tidyverse)
library(microbiome)
library(mia)
library(compositions)
library(medflex)
library(ggsci)
library(ggpubr)
导入数据
大家通过以下链接下载数据:
- 百度网盘链接:https://pan.baidu.com/s/1fz5tWy4mpJ7nd6C260abtg
- 提取码: 请关注WX公zhong号_生信学习者_后台发送 复现gmsb 获取提取码
df_v1 <- read_csv("./data/GMSB-data/df_v1.csv", show_col_types = FALSE)
bias_corr_species <- read_csv("./data/GMSB-data/results/outputs/bias_corr_species.csv")
sig_species_raw1 <- read.xlsx("./data/GMSB-data/results/outputs/res_ancombc2.xlsx", sheet = 1)
sig_species_raw2 <- read.xlsx("./data/GMSB-data/results/outputs/res_ancombc2.xlsx", sheet = 2)
# 趋势分析结果
ne_trend_test <- readRDS("./data/GMSB-data/rds/ne_trend_test.rds")
数据预处理
-
提取差异物种丰度表
-
合并分组变量和差异物种丰度表
df_v1 <- df_v1 %>%
dplyr::filter(
group1 != "missing",
druguse != "missing")
# Microbiome data
bias_corr_species <- bias_corr_species %>%
dplyr::rowwise() %>%
dplyr::filter(grepl("Species:", species)|grepl("Genus:", species)) %>%
dplyr::mutate(species = ifelse(grepl("Genus:", species),
paste(strsplit(species, ":")[[1]][2], "spp."),
strsplit(species, ":")[[1]][2])) %>%
dplyr::ungroup()
# Significant taxa by group
sig_species1 <- sig_species_raw1 %>%
dplyr::filter(p_val < 0.05) %>%
.$taxon
# Significant taxa by status
sig_species2 <- sig_species_raw2 %>%
dplyr::filter(p_statussc < 0.05) %>%
.$taxon
sig_species <- sort(base::intersect(sig_species1, sig_species2))
# Subset significant taxa
df_da_species <- bias_corr_species %>%
dplyr::filter(species %in% sig_species)
df_da_species <- t(df_da_species)
colnames(df_da_species) <- df_da_species[1, ]
df_da_species <- data.frame(df_da_species[-1, , drop = FALSE], check.names = FALSE) %>%
rownames_to_column("sampleid") %>%
dplyr::mutate(across(-1, as.numeric))
# Exposure, outcome, confounders, and potential mediators
# cytokines overlap: sCD14 and sCD163
# SCFA overlap: none
df_causal <- df_v1 %>%
dplyr::select(sampleid, recept_anal, group1, status, druguse, cd14, cd163) %>%
dplyr::left_join(df_da_species, by = "sampleid")
df_causal$status <- factor(df_causal$status)
df_causal$group1 <- factor(df_causal$group1)
df_causal$druguse <- factor(df_causal$druguse)
df_causal <- data.frame(df_causal)
head(df_causal)
| sampleid | recept_anal | group1 | status | druguse | cd14 | cd163 | Dehalobacterium.spp. | Bacteroides.spp. | ||
| 1 | F-1 | 5 | g3 | nc | yes | 1681.160 | 665.6528 | NA | 0.1151280 | |
| 2 | F-2 | 6 | g4 | nc | yes | 1178.440 | 336.1164 | -0.1870557 | -1.0903494 | |
| 3 | F-3 | 3 | g3 | nc | yes | 1717.935 | 495.9060 | NA | -0.3093994 | |
| 4 | F-4 | 7 | g4 | nc | yes | 1271.675 | 536.5375 | NA | -3.3221487 | |
| 5 | F-5 | 4 | g3 | nc | yes | 929.645 | 472.6636 | -1.1128170 | -1.2803083 | |
| 6 | F-6 | 4 | g3 | nc | no | 1103.670 | 382.0072 | NA | 1.8015313 |
函数
-
constrain_est:提取约束线性模型的beta值 -
l_infty:计算l_infty norm值 -
trend_test:趋势检验
# Estimate coefficients under constraints
constrain_est <- function(
beta_hat,
vcov_hat,
contrast,
solver = "ECOS") {
beta_opt <- CVXR::Variable(rows = length(beta_hat), cols = 1, name = "beta")
obj <- CVXR::Minimize(CVXR::matrix_frac(beta_opt - beta_hat, vcov_hat))
cons <- suppressMessages(contrast %*% beta_opt >= 0)
problem <- CVXR::Problem(objective = obj, constraints = list(cons))
suppressMessages(result <- try(CVXR::solve(problem, solver = solver), silent = TRUE))
if (inherits(result, "try-error")) {
beta_opt <- rep(0, length(beta_hat))
} else {
beta_opt <- as.numeric(result$getValue(beta_opt))
}
return(beta_opt)
}
# Compute the l_infty norm for a pattern
l_infty <- function(
beta_opt,
node) {
l <- max(abs(beta_opt[node]),
abs(beta_opt[node] - beta_opt[length(beta_opt)]),
na.rm = TRUE)
return(l)
}
# Trend test
trend_test <- function(
beta_hat,
vcov_hat,
contrast,
solver = "ECOS",
node,
B = 1000) {
beta_opt <- constrain_est(
beta_hat = beta_hat,
vcov_hat = vcov_hat,
contrast = contrast,
solver = solver)
l_opt <- l_infty(beta = beta_opt, node = node)
beta_null <- MASS::mvrnorm(
n = B,
mu = rep(0, length(beta_hat)),
Sigma = vcov_hat)
l_null <- apply(beta_null, 1, function(x) {
beta_trend <- constrain_est(beta_hat = x,
vcov_hat = vcov_hat,
contrast = contrast,
solver = solver)
l_trend <- l_infty(beta = beta_trend, node = node)
})
p_trend <- 1/B * sum(l_null > l_opt)
res <- list(estimate = beta_opt,
test_statistic = l_opt,
p_value = p_trend)
return(res)
}
contrast_mat <- matrix(
c(1, 0, 0,
-1, 1, 0,
0, -1, 1),
nrow = 3, byrow = TRUE)
Cytokine mediators
炎症细胞因子作为中介变量
All cytokines
-
多重插补
medflex::neImpute():- 使用
medflex::neImpute()函数进行多重插补。 - status是响应变量,而group1, cd14, cd163, druguse是预测变量。
- family = binomial(“logit”)指定了使用逻辑斯蒂回归模型,适用于二分类结果的变量。
- nMed = 2表示生成两个不同的插补数据集。
group1生成了group10和group11两个插补数据集。- 这个步骤是为了处理数据中的缺失值,通过生成多个完整的数据集来模拟缺失数据的可能值。
- 使用
-
构建自然效应模型
Natural effects model:- 使用
medflex::neModel()函数来拟合自然效应模型。 - 公式status ~ group10 + group11 + druguse定义了模型,其中status是响应变量,group10, group11, druguse是预测变量。
group10和group11来自medflex::neImpute()函数的多重插补。 - family = binomial(“logit”)再次指定了模型的分布族和链接函数。expData = df_exp指定了使用经过多重插补的数据集来进行模型拟合。
- se = "robust"表示使用稳健的标准误差估计,这有助于在模型估计中减少异方差性的影响。
- 使用
-
中介分析使用
Natural effects model:- exposure variables (自变量X): consisting of sexual exposure groups
- mediators (中介变量M): cytokines
- outcome variable (因变量Y): HIV-1 seroconversion status
- adjusted variable (混淆变量): druguse
- NDE: natural direct effect(自变量X不经过中介变量M直接对因变量Y的效应大小)
- NIE: natural indirect effect(自变量X仅通过中介变量M间接对因变量Y的效应大小)
df <- df_causal %>%
dplyr::select(status, group1, cd14, cd163, druguse) %>%
drop_na()
df_exp <- medflex::neImpute(
status ~ group1 + cd14 + cd163 + druguse,
family = binomial("logit"),
nMed = 2,
data = df)
ne_mod <- medflex::neModel(
status ~ group10 + group11 + druguse,
family = binomial("logit"),
expData = df_exp,
se = "robust")
summ <- summary(ne_mod)
df_summ <- data.frame(summ$coefficients)
# Trend test
# set.seed(123)
# trend_nde <- trend_test(
# beta_hat = summ$coefficients[2:4, "Estimate"],
# vcov_hat = ne_mod$vcov[2:4, 2:4],
# contrast = contrast_mat,
# node = 3, B = 1000)
#
# set.seed(123)
# trend_nie <- trend_test(
# beta_hat = summ$coefficients[5:7, "Estimate"],
# vcov_hat = ne_mod$vcov[5:7, 5:7],
# contrast = contrast_mat,
# node = 3, B = 1000)
#
# ne_trend_test <- base::append(
# ne_trend_test,
# list(cyto_nde = trend_nde, cyto_nie = trend_nie))
# Outputs
types <- c("nde", "nie")
groups <- c("g2", "g3", "g4")
res <- data.frame(
type = rep(types, each = length(groups)),
group = rep(groups, length(types)),
estimate = NA, se = NA, p = NA,
trend_p = NA)
res$estimate <- round(df_summ$Estimate[2:7], 2)
res$se <- round(df_summ$Std..Error[2:7], 2)
res$p <- round(df_summ$Pr...z..[2:7], 3)
res$trend_p[3] <- round(ne_trend_test$cyto_nde$p_value, 3)
res$trend_p[6] <- round(ne_trend_test$cyto_nie$p_value, 3)
head(res)
| type | group | estimate | se | p | trend_p | |
|---|---|---|---|---|---|---|
| 1 | nde | g2 | 1.92 | 0.64 | 0.003 | NA |
| 2 | nde | g3 | 2.56 | 0.61 | 0.000 | NA |
| 3 | nde | g4 | 3.55 | 0.70 | 0.000 | 0.000 |
| 4 | nie | g2 | 0.11 | 0.10 | 0.284 | NA |
| 5 | nie | g3 | 0.12 | 0.09 | 0.168 | NA |
| 6 | nie | g4 | 0.33 | 0.14 | 0.023 | 0.007 |
结果:炎症细胞因子cytokines在不同分组的直接和间接效应的结果
-
nde直接效应:具体来说,对于没有
druguse的受试者,增加从第1组到另一组的暴露【同时保持sCD14和sCD163在同一水平】显着增加了血清转化的几率。第2、3、4组的优势比为exp(1.92) = 6.82; Exp (2.56) = 12.94, Exp (3.55) = 34.81; -
nie间接效应,对于未
druguse的受试者,将sCD14和sCD163的水平从第1组观察到的水平转移到第4组可能看到的水平,同时在任何给定组保持暴露不变,增加血清转化的几率,比值比为exp(0.33) = 1.39。炎症因子水平从g1组水平转变成g4组,则对应HIV-1血清风险增加。
Individual cytokines
单个细胞因子的中介分析
features <- c("cd14", "cd163")
groups <- c("g2", "g3", "g4")
res_nde <- data.frame(type = "nde",
feature = rep(features, each = length(groups)),
group = rep(groups, length(features)),
estimate = NA, se = NA, p = NA)
res_nie <- data.frame(type = "nie",
feature = rep(features, each = length(groups)),
group = rep(groups, length(features)),
estimate = NA, se = NA, p = NA)
for (i in seq_along(features)) {
df <- df_causal %>%
dplyr::select(status, group1, druguse, all_of(features[i])) %>%
drop_na()
t_formula <- as.formula(paste0("status ~ group1 + ", features[i], " + druguse"))
df_exp <- neImpute(t_formula, family = binomial("logit"), data = df)
ne_mod <- neModel(status ~ group10 + group11 + druguse,
family = binomial("logit"), expData = df_exp, se = "robust")
summ <- summary(ne_mod)
idx <- seq_along(groups) + (i - 1) * length(groups)
res_nde[idx, "estimate"] <- round(summ$coefficients[2:4, "Estimate"], 2)
res_nde[idx, "se"] <- round(summ$coefficients[2:4, "Std. Error"], 2)
res_nde[idx, "p"] <- round(summ$coefficients[2:4, "Pr(>|z|)"], 3)
res_nie[idx, "estimate"] <- round(summ$coefficients[5:7, "Estimate"], 2)
res_nie[idx, "se"] <- round(summ$coefficients[5:7, "Std. Error"], 2)
res_nie[idx, "p"] <- round(summ$coefficients[5:7, "Pr(>|z|)"], 3)
}
res <- rbind(res_nde, res_nie)
head(res)
| type | feature | group | estimate | se | p | |
|---|---|---|---|---|---|---|
| 1 | nde | cd14 | g2 | 1.99 | 0.64 | 0.002 |
| 2 | nde | cd14 | g3 | 2.59 | 0.61 | 0.000 |
| 3 | nde | cd14 | g4 | 3.66 | 0.69 | 0.000 |
| 4 | nde | cd163 | g2 | 2.01 | 0.65 | 0.002 |
| 5 | nde | cd163 | g3 | 2.69 | 0.62 | 0.000 |
| 6 | nde | cd163 | g4 | 3.76 | 0.71 | 0.000 |
Microbial species
炎症细胞因子作为中介变量
All species
-
中介分析使用
Natural effects model:- exposure variables (自变量X): consisting of sexual exposure groups
- mediators (中介变量M): gut microbiota
- outcome variable (因变量Y): HIV-1 seroconversion status
- adjusted variable (混淆变量): druguse
# Natural effects model
all_species <- colnames(df_causal)[8:16]
df <- df_causal %>%
dplyr::select(status, group1, druguse, all_of(all_species))
df[is.na(df)] <- 0
t_formula <- as.formula(paste0("status ~ group1 + ",
paste0(all_species, collapse = " + "),
" + druguse"))
df_exp <- neImpute(
t_formula,
family = binomial("logit"),
nMed = length(all_species),
data = df)
ne_mod <- neModel(
status ~ group10 + group11 + druguse,
family = binomial("logit"),
expData = df_exp,
se = "robust")
summ <- summary(ne_mod)
df_summ <- data.frame(summ$coefficients)
# Trend test
# set.seed(123)
# trend_nde <- trend_test(beta_hat = summ$coefficients[2:4, "Estimate"],
# vcov_hat = ne_mod$vcov[2:4, 2:4],
# contrast = contrast_mat,
# node = 3, B = 1000)
# set.seed(123)
# trend_nie <- trend_test(beta_hat = summ$coefficients[5:7, "Estimate"],
# vcov_hat = ne_mod$vcov[5:7, 5:7],
# contrast = contrast_mat,
# node = 3, B = 1000)
#
# ne_trend_test <- base::append(ne_trend_test,
# list(species_nde = trend_nde, species_nie = trend_nie))
# Outputs
type <- c("nde", "nie")
groups <- c("g2", "g3", "g4")
res <- data.frame(type = rep(types, each = length(groups)),
group = rep(groups, length(type)),
estimate = NA, se = NA, p = NA,
trend_p = NA)
res$estimate <- round(df_summ$Estimate[2:7], 2)
res$se <- round(df_summ$Std..Error[2:7], 2)
res$p <- round(df_summ$Pr...z..[2:7], 3)
res$trend_p[3] <- round(ne_trend_test$species_nde$p_value, 3)
res$trend_p[6] <- round(ne_trend_test$species_nie$p_value, 3)
head(res)
| type | group | estimate | se | p | trend_p | |
|---|---|---|---|---|---|---|
| 1 | nde | g2 | 2.08 | 0.68 | 0.002 | NA |
| 2 | nde | g3 | 2.61 | 0.64 | 0.000 | NA |
| 3 | nde | g4 | 3.58 | 0.71 | 0.000 | 0.000 |
| 4 | nie | g2 | 0.02 | 0.13 | 0.879 | NA |
| 5 | nie | g3 | 0.15 | 0.13 | 0.264 | NA |
| 6 | nie | g4 | 0.35 | 0.17 | 0.045 | 0.033 |
Individual species
features <- sort(all_species)
groups <- c("g2", "g3", "g4")
res_nde <- data.frame(type = "nde",
feature = rep(features, each = length(groups)),
group = rep(groups, length(features)),
estimate = NA, se = NA, p = NA)
res_nie <- data.frame(type = "nie",
feature = rep(features, each = length(groups)),
group = rep(groups, length(features)),
estimate = NA, se = NA, p = NA)
for (i in seq_along(features)) {
df <- df_causal %>%
dplyr::select(status, group1, druguse, all_of(features[i])) %>%
drop_na()
t_formula <- as.formula(paste0("status ~ group1 + ", features[i], " + druguse"))
df_exp <- neImpute(t_formula, family = binomial("logit"), data = df)
ne_mod <- neModel(status ~ group10 + group11 + druguse,
family = binomial("logit"), expData = df_exp, se = "robust")
summ <- summary(ne_mod)
idx = seq_along(groups) + (i - 1) * length(groups)
res_nde[idx, "estimate"] <- round(summ$coefficients[2:4, "Estimate"], 2)
res_nde[idx, "se"] <- round(summ$coefficients[2:4, "Std. Error"], 2)
res_nde[idx, "p"] <- round(summ$coefficients[2:4, "Pr(>|z|)"], 3)
res_nie[idx, "estimate"] <- round(summ$coefficients[5:7, "Estimate"], 2)
res_nie[idx, "se"] <- round(summ$coefficients[5:7, "Std. Error"], 2)
res_nie[idx, "p"] <- round(summ$coefficients[5:7, "Pr(>|z|)"], 3)
}
res <- rbind(res_nde, res_nie)
head(res)
| type | feature | group | estimate | se | p | |
|---|---|---|---|---|---|---|
| 1 | nde | A.muciniphila | g2 | -0.62 | 1.37 | 0.649 |
| 2 | nde | A.muciniphila | g3 | 1.57 | 0.88 | 0.076 |
| 3 | nde | A.muciniphila | g4 | 3.46 | 1.37 | 0.012 |
| 4 | nde | B.caccae | g2 | 0.90 | 1.03 | 0.382 |
| 5 | nde | B.caccae | g3 | 2.09 | 0.95 | 0.028 |
| 6 | nde | B.caccae | g4 | 3.28 | 1.20 | 0.006 |
Combine cytokines and DA species
-
中介分析使用
Natural effects model:- exposure variables (自变量X): consisting of sexual exposure groups
- mediators (中介变量M): cytokines & gut microbiota
- outcome variable (因变量Y): HIV-1 seroconversion status
- adjusted variable (混淆变量): druguse
# Natural effects model
all_mediators <- c(all_species, "cd14", "cd163")
df <- df_causal %>%
dplyr::select(status, group1, druguse, all_of(all_mediators))
df[is.na(df)] <- 0
t_formula <- as.formula(paste0("status ~ group1 + ",
paste0(all_mediators, collapse = " + "),
" + druguse"))
df_exp <- neImpute(
t_formula,
family = binomial("logit"),
nMed = length(all_mediators),
data = df)
ne_mod <- neModel(
status ~ group10 + group11 + druguse,
family = binomial("logit"),
expData = df_exp,
se = "robust")
summ <- summary(ne_mod)
df_summ <- data.frame(summ$coefficients)
# Trend test
# set.seed(123)
# trend_nde <- trend_test(beta_hat = summ$coefficients[2:4, "Estimate"],
# vcov_hat = ne_mod$vcov[2:4, 2:4],
# contrast = contrast_mat,
# node = 3, B = 1000)
# set.seed(123)
# trend_nie <- trend_test(beta_hat = summ$coefficients[5:7, "Estimate"],
# vcov_hat = ne_mod$vcov[5:7, 5:7],
# contrast = contrast_mat,
# node = 3, B = 1000)
#
# ne_trend_test <- base::append(ne_trend_test,
# list(all_nde = trend_nde, all_nie = trend_nie))
# Outputs
types <- c("nde", "nie")
groups <- c("g2", "g3", "g4")
res <- data.frame(type = rep(types, each = length(groups)),
group = rep(groups, length(types)),
estimate = NA, se = NA, p = NA,
trend_p = NA)
res$estimate <- round(df_summ$Estimate[2:7], 2)
res$se <- round(df_summ$Std..Error[2:7], 2)
res$p <- round(df_summ$Pr...z..[2:7], 3)
res$trend_p[3] <- round(ne_trend_test$all_nde$p_value, 3)
res$trend_p[6] <- round(ne_trend_test$all_nie$p_value, 3)
head(res)
| type | group | estimate | se | p | trend_p | |
|---|---|---|---|---|---|---|
| 1 | nde | g2 | 1.81 | 0.64 | 0.005 | NA |
| 2 | nde | g3 | 2.38 | 0.61 | 0.000 | NA |
| 3 | nde | g4 | 3.16 | 0.68 | 0.000 | 0.000 |
| 4 | nie | g2 | 0.20 | 0.17 | 0.241 | NA |
| 5 | nie | g3 | 0.29 | 0.17 | 0.087 | NA |
| 6 | nie | g4 | 0.74 | 0.24 | 0.002 | 0.001 |
Additional analysis: treating the exposure as continuous
-
中介分析使用
Natural effects model:- exposure variables (自变量X): recept_anal
- mediators (中介变量M): cytokines & gut microbiota
- outcome variable (因变量Y): HIV-1 seroconversion status
- adjusted variable (混淆变量): druguse
# Natural effects model
all_mediators <- c(all_species, "cd14", "cd163")
df <- df_causal %>%
dplyr::select(status, recept_anal, druguse, all_of(all_mediators))
df[is.na(df)] <- 0
t_formula <- as.formula(paste0("status ~ recept_anal + ",
paste0(all_mediators, collapse = " + "),
" + druguse"))
df_exp <- neImpute(t_formula,
family = binomial("logit"), nMed = length(all_mediators), data = df)
ne_mod <- neModel(status ~ recept_anal0 + recept_anal1 + druguse,
family = binomial("logit"), expData = df_exp, se = "robust")
summ <- summary(ne_mod)
df_summ <- data.frame(summ$coefficients)
df_summ
| Estimate | Std…Error | z.value | Pr…z… | |
|---|---|---|---|---|
| (Intercept) | -1.92892873 | 0.419607493 | -4.596984 | 4.286515e-06 |
| recept_anal0 | 0.08028533 | 0.040929531 | 1.961550 | 4.981487e-02 |
| recept_anal1 | 0.00831693 | 0.004785597 | 1.737909 | 8.222692e-02 |
| druguseyes | 1.17852596 | 0.434863654 | 2.710105 | 6.726201e-03 |
Additional analysis: substance usage as the mediator
-
中介分析使用
Natural effects model:-
exposure variables (自变量X): recept_anal
-
mediators (中介变量M): druguse
-
outcome variable (因变量Y): HIV-1 seroconversion status
-
# Natural effects model
df <- df_causal %>%
dplyr::select(status, group1, druguse)
df[is.na(df)] <- 0
t_formula <- as.formula(paste0("status ~ group1 + druguse"))
df_exp <- neImpute(t_formula,
family = binomial("logit"), data = df)
ne_mod <- neModel(status ~ group10 + group11,
family = binomial("logit"), expData = df_exp, se = "robust")
summ <- summary(ne_mod)
df_summ <- data.frame(summ$coefficients)
df_summ
| Estimate | Std…Error | z.value | Pr…z… | |
|---|---|---|---|---|
| (Intercept) | -3.01328264 | 0.59597082 | -5.056091 | 4.279374e-07 |
| group10g2 | 2.07838397 | 0.65668493 | 3.164964 | 1.551023e-03 |
| group10g3 | 2.71549102 | 0.62579799 | 4.339245 | 1.429728e-05 |
| group10g4 | 3.88678487 | 0.70600884 | 5.505292 | 3.685567e-08 |
| group11g2 | 0.07609705 | 0.07185271 | 1.059070 | 2.895679e-01 |
| group11g3 | 0.18057639 | 0.11016834 | 1.639095 | 1.011934e-01 |
| group11g4 | 0.17817557 | 0.12483109 | 1.427333 | 1.534839e-01 |

















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











