




 Picard是一个用于处理排序数据如SAM/BAM/CRAM和VCF文件的命令行工具。它依赖Java环境并提供如CollectInsertSizeMetrics的功能,用于统计插入片段大小分布。这个工具对于生物信息学分析至关重要,尤其是DNA测序数据的处理和比对结果的可视化。
Picard是一个用于处理排序数据如SAM/BAM/CRAM和VCF文件的命令行工具。它依赖Java环境并提供如CollectInsertSizeMetrics的功能,用于统计插入片段大小分布。这个工具对于生物信息学分析至关重要,尤其是DNA测序数据的处理和比对结果的可视化。
下游比对数据的统计工具 picard
Picard是一组命令行工具,用于处理高通量排序数据和格式,如sam/ bam/ cran和vcf文件。
安装
在Linux系统目录下执行以下命令下载软件
wget https://github.com/broadinstitute/picard/releases/download/2.25.5/picard.jar
Picard 官网
http://broadinstitute.github.io/picard/
运行
软件依赖于java环境,可通过以下命令查看Java是否安装及版本
java -version
在软件下载的目录下执行以下命令运行软件,出现下方表示软件安装成功
java -jar picard.jar
使用示例
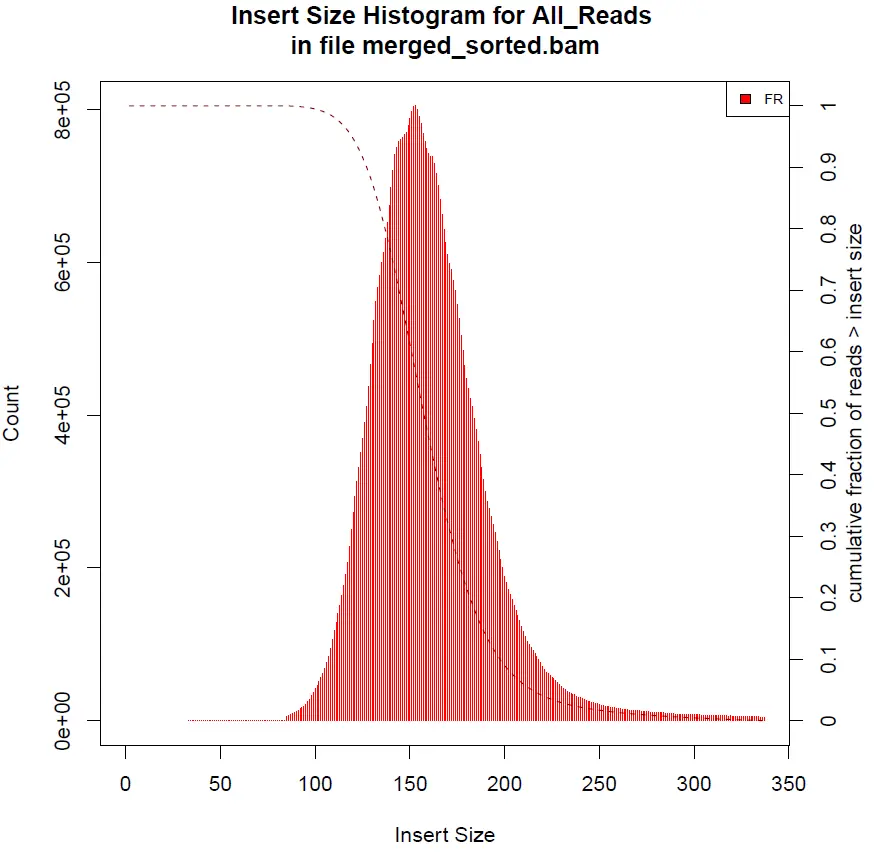
插入片段分布统计工具CollectInsertSizeMetrics
通过检测双端序列在参考基因组上的起止位置,可以得到样品DNA打断后得到的测序片段的实际大小,即插入片段大小(Insert Size),它是信息分析时的一个重要参数。插入片段大小的分布一般符合正态分布,且只有一个单峰,Insert Size分布图可以展示各个样品的插入片段的长度分布情况。
java -jar picard.jar CollectInsertSizeMetrics \
I=sorted.mkdup.bam \ # 比对后取出pcr重复后的bam文件
O=insert_size_metrics.txt \ # 输出txt文件
H=insert_size_histogram.pdf \ls # 输出含统计图的Pdf文件
M = 0.5




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











