







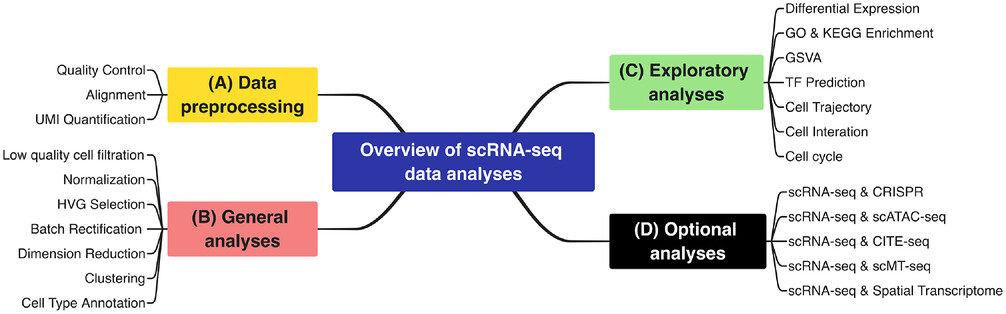
 本文详细介绍了单细胞RNA测序技术的工作流程,包括数据格式转换、比对、质控、归一化、高变基因检测、批次效应校正、降维与聚类、细胞注释、功能分析、转录因子推断和联合分析等关键步骤,展示了这一技术在揭示细胞异质性和功能多样性方面的应用。
本文详细介绍了单细胞RNA测序技术的工作流程,包括数据格式转换、比对、质控、归一化、高变基因检测、批次效应校正、降维与聚类、细胞注释、功能分析、转录因子推断和联合分析等关键步骤,展示了这一技术在揭示细胞异质性和功能多样性方面的应用。
scRNA-seq工作流程
单细胞RNA测序(scRNA-seq)技术已成为解开单个细胞内RNA转录物的异质性和复杂性,以及揭示高度组织化组织/器官/生物体内不同细胞类型和功能的组成的最先进方法。通过单细胞RNA测序,现在可以在一项研究中分析超过数百万个细胞的单细胞水平的转录组。这使我们能够在转录组水平上对每个细胞进行分类、表征和区分,从而识别出稀有但功能重要的细胞群。
单细胞转录组原始数据格式
数据格式一:FASTQ
FASTQ文件可以直接通过FastQC进行质控分析。
数据格式一:BCL
对于BCL格式可以使用10x工具cellranger mkfastq,将BCL文件转换为FASTQ文件,转换需要提供csv矩阵文件(包含lane、sample和index三列数据),转换之后的FASTQ文件同样可采用FastQC进行质控。
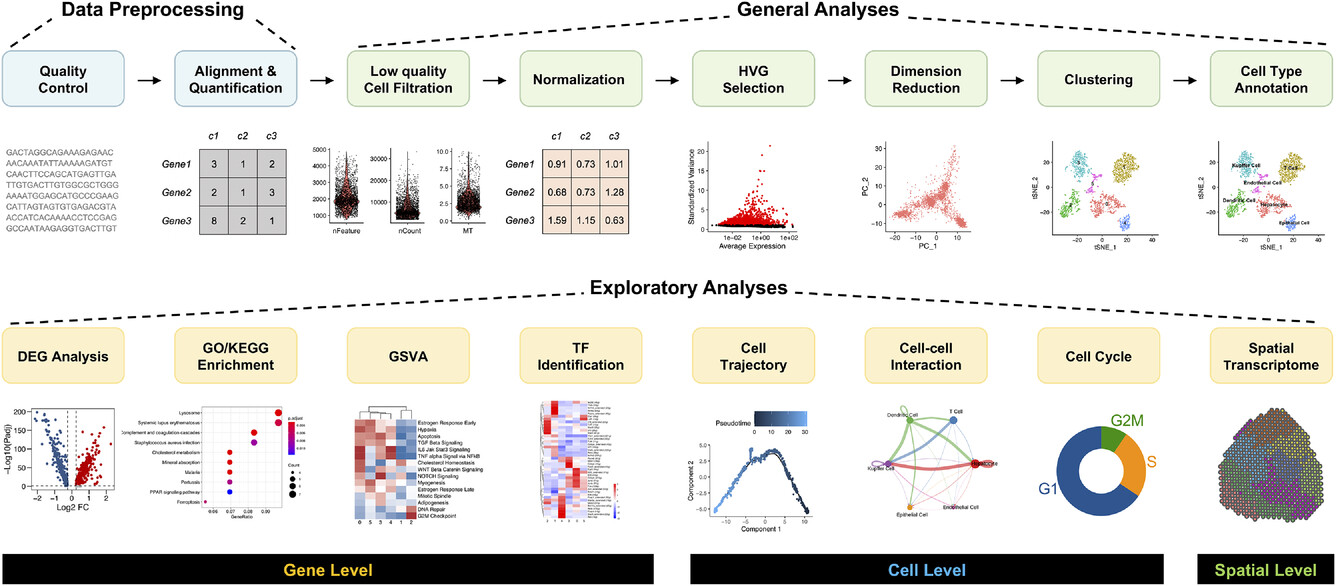
单细胞RNA-seq数据分析总览
1. 比对到参考基因组
使用STAR或Tophat等比对软件,可以将FASTQ数据比对到特定的参考基因组,cellranger使用STAR作为比对软件,然后通过基因注释GTF文件根据reads的比对情况,将reads分类为外显子、内含子和基因间区。
2. 质控
为什么要进行质控?
在单细胞悬液的制备过程中,由于实验操作、技术问题和不可避免的自然现象,可能会发生活细胞死亡,细胞膜损伤或多细胞粘附等情况。因此,为了消除低质量细胞对基因表达的干扰,需要进行质控。
质控软件
主要有Seurat、scran和scanpy,其中在引用方面,Seurat是最受欢迎的,它内置了处理低质量细胞过滤的功能。
质控指标
可以通过以下质控指标来判断:
1. 基因的数量
2. UMI(转录本)的数量
3. 线粒体基因的百分比
4. 核糖体蛋白基因在每个细胞中的百分比
过滤阈值的设置没有绝对的标准,它通常取决于被分析的细胞和组织的类型。例如过滤掉≤100或≥6000个表达基因、≤200个UMIs和≥10%线粒体基因的细胞,也可灵活调整。对于线粒体基因的细胞过滤,对于心肌细胞等类型细胞,需要谨慎处理。
3. 数据归一化
归一化旨在抵消技术噪声或偏差,并确保每个细胞之间的可比性。
与分析传统的bulk RNA-seq数据类似,在分析单细胞RNA测序数据时,每个细胞都被视为一个独立的样本。原始表达矩阵不能直接用于下游分析,因为由于系统错误或技术噪音(如每个细胞的测序深度和转录组捕获率的差异),细胞之间的表达水平无法进行比较。
归一化工具
归一化工具有BASiCS、GRM、Linnorm、SAMstrt、SCnorm、scran和Simple norm等,其中innorm和scran的速度优势来自于用C 编写,并在R中实现,适合于大数据集。相反,BASiCS和SCnorm需要更长的时间来生成更精确的结果。总的来说,这些方法之间存在很大的差异,不同的工具在不同的情况下表现最佳。
4. 检测高变基因(HGVS)
单细胞RNA测序数据集是高维的,一个样本中有数万个细胞,每个细胞中有数千个基因表达。每个细胞中的大部分基因都属于家族基因,因为它们的特点是细胞之间的表达水平没有显著变化,它们的存在往往会掩盖真正的生物信号。
什么是高变基因?
在数据集中表现出高度细胞间变异的特征子集也被称为高度可变基因(HVGs)。HVGs不仅突出了生物信号,而且由于计算量的显著减少,大大加快了对单细胞RNA测序数据下游分析的速度。一个高质量的HVGs应该包含能够区分不同细胞类型的基因,HVGs的质量对聚类的精度有显著影响。
检测高变基因工具
主要有BASiCS、Brennecke、scLVM、scran、scVEGs和Seurat等,其中scran可以检测出稳定数量的HVGs,并且运行速度较快。Brennecke在大范围数据集上具有稳定一致的性能。scran和Seurat在处理部分数据集时表现最佳。
5. 批次效应校正
为什么要校正批次效应?
不同的单细胞数据可能产生于不同的时间、不同的测序平台,这些数据之间不可避免地存在着技术上或非生物学上的显著批次效应。如果不进行校正,它可以匹配基因表达模式,进而导致错误的结论。
批次效应校正方法
有Harmony、Scanorama、Seurat V4以及基于深度学习的deepMNN等方法,deepMNN在精度优于现有的常用方法,特别是在大数据集的情况下。
6. 降维
降维是处理单细胞高维数据的主要策略之一。对于单细胞RNA测序数据,通常需要进行2次降维处理,分别为主成分分析(PCA)降维和t-SNE/UMAP降维,并进行可视化。
PCA
PCA是一种数学线性维度算法,它利用正交变换将一系列可能线性相关的变量转换为新的线性不相关的变量,从而利用新的变量在低维上显示数据的特征。PCA已广泛应用于scRNA-seq研究,以克服任何单一特征中的噪音。
t-SNE/UMAP
UMAP在细胞子集的连续性方面比t-SNE有明显的优势,因为其保留了更多的全局结构,但t-SNE由于良好的可视化效果也应用于许多单细胞研究。
7. 聚类
单细胞RNA测序数据的复杂性促进的聚类方法的应用。
聚类方法
SC3和Seurat综合来看表现更好,其中Seurat的分析速度要更快。在cluster数量相同的情况下,Seurat通常与真实情况的一致性最好,而FlowSOM在cluster数量大于真实数量时其与真实情况的一致性更好。
8. 注释
在scRNA-seq数据中注释细胞的工作流程包括三个主要步骤:自动注释、人工注释和湿实验验证。
自动注释
自动化注释工具利用一组预定义的标记基因,这些标记基因在已知的细胞类型中特异表达,通过将它们的基因表达模式与已知的细胞类型进行匹配来标记cluster。优点是快速、可重复性好,对常见细胞类型的标注结果更可靠; 但由于参考标记基因集的限制,它无法定义罕见的和新的细胞类型。
在Seurat、SingleR、CP、RPC和SingleCellNet这五种最常用的方法中,Seurat是注释主要细胞类型的效果较好, 但Seurat在预测罕见细胞类型和区分高度相似的细胞类型方面表现相对较差。
人工注释
人工注释是标注细胞的金标准,虽然它需要搜索相关文献和挖掘已有的scRNA-seq数据,较为主观并且耗时。
湿实验验证
通常需要湿实验来进一步验证scRNA-seq的发现。传统的验证方法包括免疫荧光和免疫组化,这两种方法都是基于抗体与抗原(标记基因编码的表面蛋白)特异性结合的原理来证明数据分析得到的细胞类型的真实存在。
9. 功能富集分析
为了准确地揭示特定细胞群体的功能和生物学意义,有必要对目标差异表达基因集进行功能富集分析。功能富集的通用分析策略也适用于单细胞数据,如GO和KEGG通路。此外,GSVA以通路为中心的方式广泛应用于功能富集分析等标准分析中。GSVA可以计算每个样本中不同信号通路的富集分数,以评估表型差异的原因,可以作为KEGG通路的补充,使结果更具生物学解释性。
10. 转录因子(TF)的推断
为了从scRNA-seq数据中识别每个细胞簇中富集的转录因子,可使用SCENIC软件实现转录因子(TF)的推断,主要原理是:通过搜索靶基因的假定的调控区域来富集转录因子基序,然后转录因子基序富集可以实现候选TF调控因子与候选靶基因的连接。SCENIC有R和Python版本,推荐使用pySCENIC运行大的数据集,当前支持人、小鼠和果蝇等物种,也可以手动创建其他物种的定制数据库。
11. 拟时分析
拟时分析可以在单细胞水平上推断细胞的轨迹,有望发现罕见的细胞类型和隐蔽的状态。Monocle是应用最广泛的拟时分析工具之一,它借鉴显式主图来描述数据,并通过嵌入反向图来重建单细胞轨迹,以提高预测轨迹的稳健性和准确性。
12. 细胞-细胞通讯
生物体受到刺激后会自我调节以维持体内稳态,这需要多种细胞的共同参与和协调。随着细胞-细胞通讯研究的快速发展,出现了诸如CellChat、CellPhoneDB、NicheNet、SingleCellSignalR和iTalk等分析工具,每一种工具都依赖于细胞表面配体和受体相互作用的强度,但每一种工具都有其优缺点。具体来说,如果要考虑配体和受体的结构组成,CellPhoneDB是首选。如果需要考虑辅助因子(如启动子和拮抗剂)的调控,可以选择CellChat来提高性能。
13. 细胞周期阶段预测
单细胞悬液中的每个细胞都处于细胞周期的特定阶段:DNA合成前期(G1期)、DNA合成期(S期)、DNA合成后期(G2期)或有丝分裂期(M期),每群细胞都混合着不同细胞周期的细胞。Seurat的CellCycleScoring功能可根据其内置包内的G2/M和S期标记基因的表达给每个细胞打分。
14. 联合分析
如scRNA-seq和CRISPR筛选的联合应用,scRNA-seq和多组学的综合分析,包括scATAC-sEquation(单细胞染色质可及性和转录组测序)、scMT-sEquation(单细胞甲基化组和转录组测序)、CITE-sEquation(通过测序对转录组和表位进行细胞索引)和空间转录组。这些技术的结合可以更好、更深入地了解关键的生物过程和机制,是未来单细胞技术发展的一个重要方向。



















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











