






记录一次宏基因组数据处理,没想到在序列比对还能踩一坑,debug硬控了一段时间。感谢万能的国外论坛老哥,给出了简单有效的解决方法。
错误信息:
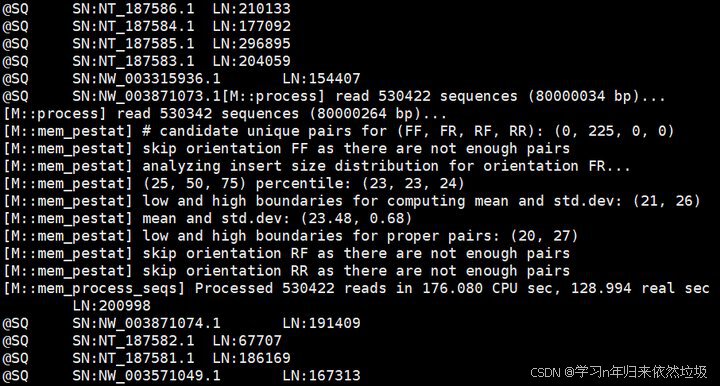
主要是sam文件格式错误,导致samtools无法读取。查看sam文件,包含了bwa的日志信息[M::bwa_idx_load_from_disk] read 0 ALT contigs,[M::process] read 530342 sequences (80000264 bp)...等。
背景:
拿到了一批宏基因组的双端测序数据,虽然公司已经跑过一遍,提供了在线分析报告。但还是打算自己重新跑一遍。在跟着公司的pipeline跑数据时,在序列比对就出了问题。
使用了以下脚本来处理质控后的Cleandata。错误的根源,并且在debug时反复检查觉得没有问题,导致之后error一直无法解决。
#!/bin/bash
#SBATCH --job-name=bwa_host_removal # 作业名称
#SBATCH --partition=cpuPartition # 分区名称
#SBATCH --nodes=1 # 一个节点
#SBATCH --ntasks-per-node=8 # 每个节点使用8个任务
#SBATCH --error=bwa_%j.err # 错误日志文件
#SBATCH --output=bwa_%j.out # 输出日志文件
# 激活 Conda 环境
source ~/miniconda3/bin/activate metageno
# 输入和输出目录
INPUT_DIR="~/cleandata"
OUTPUT_DIR="~/cleandata_no_host"
REFERENCE="~/index/index"
# 确保输出目录存在
mkdir -p ${OUTPUT_DIR}
# 批量处理所有样本
for SAMPLE in ${INPUT_DIR}/*_R1.clean.fastq.gz; do
BASENAME=$(basename ${SAMPLE} _R1.clean.fastq.gz)
R1="${INPUT_DIR}/${BASENAME}_R1.clean.fastq.gz"
R2="${INPUT_DIR}/${BASENAME}_R2.clean.fastq.gz"
SAM="${OUTPUT_DIR}/${BASENAME}.aligned.sam"
CLEAN_R1="${OUTPUT_DIR}/${BASENAME}_R1.no_host.fastq.gz"
CLEAN_R2="${OUTPUT_DIR}/${BASENAME}_R2.no_host.fastq.gz"
# 比对到参考基因组
bwa mem -t 8 ${REFERENCE} ${R1} ${R2} > ${SAM}
# 去除宿主序列生成无污染的fastq
samtools view -bS ${SAM} | samtools fastq -f 4 -1 ${CLEAN_R1} -2 ${CLEAN_R2}
# 删除临时sam文件
rm ${SAM}
done
# 停止conda环境
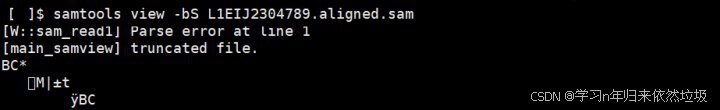
conda deactivate脚本在调用samtools时开始报错,sam文件已损坏。单独调用samtools查看文件,确实存在格式问题。
查看了sam文件大小,看起来似乎正常,并且并且各样本间差异不大。
使用以下命令查看了sam文件的内容,得到了开始时的error信息。
# 查看sam文件前560行内容
head -n 560 L1EIJ2304789.aligned.sam看到第1行[M::bwa_idx_load_from_disk] read 0 ALT contigs,以及第545行开始[M::process] read 530342 sequences (80000264 bp)...。sam 文件中出现了 bwa 的日志信息。
之后重新去下载了参考基因组生成索引。按照脚本的代码,单独调用bwa mem重新尝试运行了几次,结果都没变。
解决方案
不得不感谢国外论坛老哥,给出非常有效的解决方法。终于发现是因为生成sam文件时使用了重定向(>)的原因,似乎把bwa运行的stderr结果也输出到sam文件中了。
解决方法也很简单,可以使用管道把bwa的输出结果直接传给samtools。运行后生成了正常的bam文件,也能正常去除比对上的reads。
# 使用 bwa mem 进行比对并将结果转换为 BAM 格式
bwa mem -t 8 index ~/cleandata/L1EIJ2304789_R1.clean.fastq.gz ~/cleandata/L1EIJ2304789_R2.clean.fastq.gz | samtools view -Sb - > ~/cleandata_no_host/L1EIJ2304789.bam
#从 BAM 文件中提取无宿主序列
samtools fastq -f 4 -1 ~/nohost_fastq/L1EIJ2304789_R1.no_host.fastq.gz -2 ~/nohost_fastq/L1EIJ2304789_R2.no_host.fastq.gz ~/cleandata_no_host/L1EIJ2304789.bam
此外,在论坛上似乎有因为使用nohup挂起运行bwa导致同样的sam文件格式错误的情况,不使用nohup后问题解决。在debug时,单独运行bwa时同样使用了nohup,结果还是错误格式的sam文件,不确定在这个错误里是否有nohup的原因。但由于sam文件太大bwa运行时间久,暂时没有尝试这种解决方案。
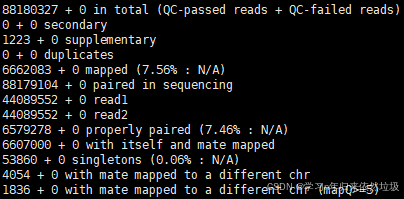
验证宿主序列去除效果,使用samtools flagstat统计文件的比对情况。
samtools flagstat ~/cleandata_no_host/L1EIJ2304789.bam
7.56%的比对率,有92.44% 的 reads 没有比对到宿主,和公司报告的92.86%非常接近。可能由于使用的bwa和samtools版本不同,或者部分阈值设置不同导致。这些差异在统计时通常是可以接受的,于是问题基本解决。

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









