







今天介绍的是发表在biorxiv上的一篇名为“High-accuracy protein complex structure modeling based on sequence-derived structure complementarity”的文章。确定蛋白质复合物结构对理解生物功能至关重要。虽然AlphaFold2在单体蛋白结构预测上取得突破,但捕捉链间相互作用信号和建模复合物结构仍具挑战。本文介绍DeepSCFold模型,用于改进蛋白质复合物建模,利用基于序列的深度学习模型预测结构相似性和相互作用概率,为构建配对多序列比对(MSAs)提供基础。基准测试显示,DeepSCFold显著优于现有方法:在CASP15多聚体目标中,TM-score分别比AlphaFold-Multimer和AlphaFold3提高11.6%和10.3%;在SAbDab抗体-抗原复合物中,结合界面预测成功率分别提升24.7%和12.4%。结果表明,DeepSCFold通过序列衍生信息有效捕获保守的蛋白质相互作用模式,减少对序列级别的共进化信号的依赖。
1.引言
蛋白质通过相互作用形成多蛋白复合物(multimers或assemblies),在细胞信号转导、物质运输和代谢等关键生物过程中发挥核心作用。然而,解析这些复合物的三维结构对于理解其功能至关重要,但实验技术(如X射线晶体学、核磁共振和冷冻电镜)往往面临分辨率、时间和成本上的限制。因此,获得复杂结构的计算方法已经成为结构生物学实验技术的不可或缺的重要补充。
传统预测方法包括基于模板的同源建模和分子对接方法。前者受限于高质量模板的稀缺性,后者则因构象采样复杂性和界面柔性等问题难以准确预测复合物结构。近年来,深度学习方法(如AlphaFold2及其扩展版本AlphaFold-Multimer)在单体蛋白结构预测上取得革命性进展,但在复合物预测中仍存在显著差距,尤其是对于缺乏序列共进化信号的系统(如抗体-抗原复合物)。
多序列比对(MSAs)是蛋白质复合物结构预测的关键,其质量直接影响预测准确性。在蛋白质复合物中,准确捕捉蛋白质链之间的结合模式可能显著受益于复合物的配对MSAs。然而,现有工具(如HHblits、Jackhammer和MMseqs)主要用于构建单体MSAs,无法直接生成配对MSAs,这限制了对链间相互作用的捕捉能力。尽管一些新方法(如DeepMSA、MULTICOM和DiffPALM)试图解决这一问题,但它们在缺乏序列共进化信号的复杂系统中仍显不足。
值得注意的是,蛋白质结构通常比序列更保守,尤其是在蛋白质-蛋白质相互作用(PPIs)界面上。实验证据表明,自然界中蛋白质相互作用模式的多样性非常有限,不同的PPIs中观察到类似的结构结合模式。这种保守性为开发基于结构互补性的新方法提供了理论基础。为此,我们开发了DeepSCFold——一种整合序列语义嵌入和理化特征的深度学习框架。DeepSCFold不依赖序列共进化信号,而是通过探索空间构象互补性全面捕捉链间相互作用,从而显著提升预测精度。
在CASP15和SAbDab数据库上的基准测试表明,DeepSCFold在全局和局部界面准确性方面均优于现有最先进方法。尤其是在缺乏链间共进化信号的抗体-抗原复合物中,DeepSCFold通过基于结构互补性的配对MSAs有效弥补了信息缺失,展现了卓越的鲁棒性和预测能力。
2.方法
2.1 模型架构
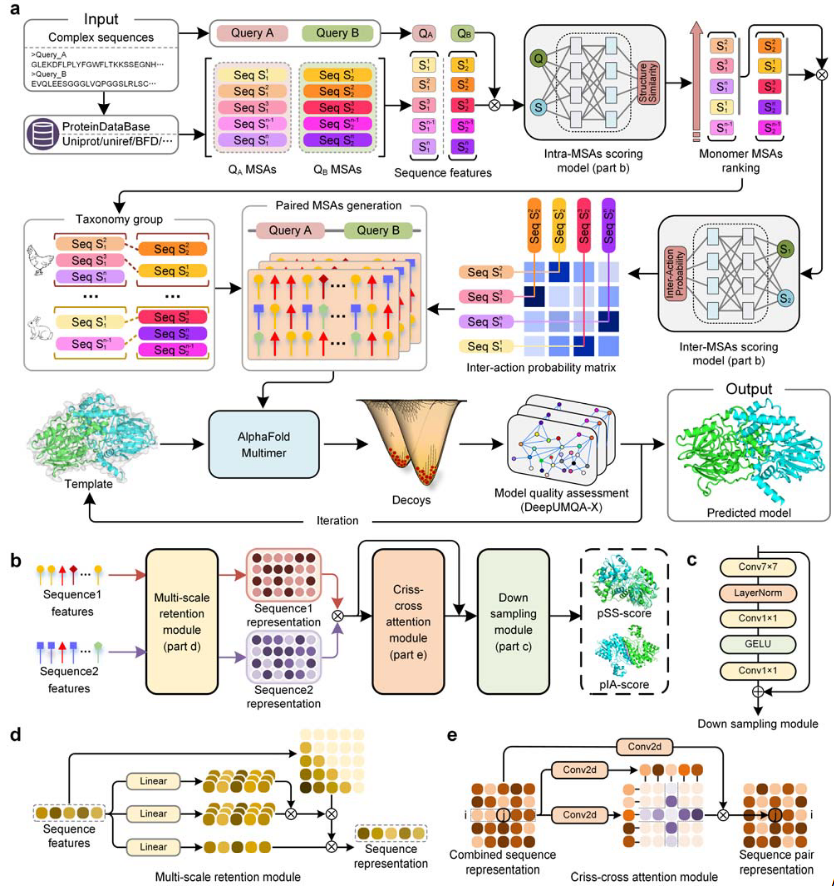
图1 DeepSCFold的流程
DeepSCFold的核心目标是进行高精度蛋白质复合物结构预测,其核心在于通过深度学习和多序列比对(MSAs)技术捕捉蛋白质链间的结构互补性和相互作用信号。
构建配对多序列比对(pMSAs):
- 结构相似性评估:通过评估单体查询序列与其在单个MSAs中的对应同源物之间的结构相似性。
- 相互作用模式识别:识别不同单体MSAs中序列之间的潜在相互作用模式。
- 双重策略:这两种策略共同系统地生成高质量的pMSAs,为准确的蛋白质复合物建模提供基础。
深度学习模型:
- 结构相似性预测模型:仅从序列信息中预测蛋白质-蛋白质的结构相似性(pSS-score)。
- 相互作用概率预测模型:仅基于序列级特征估计相互作用概率(pIA-score)。
- 无需先验结构知识:这些模型能够在没有先验结构知识的情况下推断结构和相互作用特性,使DeepSCFold能够仅从序列数据中建模复杂的相互作用。
具体流程如图1a所示:首先,输入蛋白质复合物的多个查询序列,从多个序列数据库生成单体MSAs,并利用结构相似性预测模型(pSS-score)量化输入序列与同源序列之间的相似性,优化MSAs的排序;其次,通过相互作用概率预测模型(pIA-score)评估不同链间的潜在相互作用模式,构建高质量的配对MSAs,同时整合物种注释、UniProt登录号等多源生物学信息以增强相关性;最后,基于构建的配对MSAs,使用AlphaFold-Multimer进行复合物结构预测,并通过内部质量评估方法(DeepUMQA-X)选择最优模型进行迭代优化,生成最终结构。
2.2 结构相似性和相互作用概率预测
配对多序列比对(paired-MSA)对于蛋白质复合物结构建模至关重要。仅依赖传统序列相似性和现有复合物结构信息的方法在构建高质量的配对MSAs时可能面临困难。为了高效地构建配对MSAs,DeepSCFold开发了一个基于序列的深度学习模型,用于捕捉序列之间的关系。
模型的工作流程如图1b,1c,1d和1e所示:首先,提取序列衍生特征并整合来自蛋白质语言模型(如ESM2)的嵌入表示;其次,利用多尺度保留模块捕捉序列中的长程依赖关系和局部特征,生成序列表示;接着,通过交叉注意力模块捕捉两个序列间的相互作用,生成配对序列表示;最后,将配对的序列表示通过下采样模块,以预测结构相似性分数(pSS-score)或相互作用概率分数(pIA-score)。
2.3 MSA采样和配对MSA构建
为了生成高质量的多序列比对(MSAs),DeepSCFold采用多数据库搜索策略,整合UniRef30、UniRef90、UniProt50、BFD、MGnify和ColabFold等序列数据库,确保单体MSAs的广泛覆盖。在此基础上,利用预测的结构相似性分数(pSS-score)补充传统序列相似性,优化MSAs的排名和选择。
对于多聚体,通过深度学习模型预测不同亚基MSAs之间的相互作用概率(pIA-score),并基于这些概率构建配对MSAs。此外,DeepSCFold还整合物种注释、UniProt登录号及PDB中的蛋白质复合物信息,进一步丰富配对MSAs的多样性,避免局部最优,从而显著提升蛋白质复合物结构预测的准确性。
3.实验
3.1 DeepSCFold对蛋白质复合物结构预测的改进
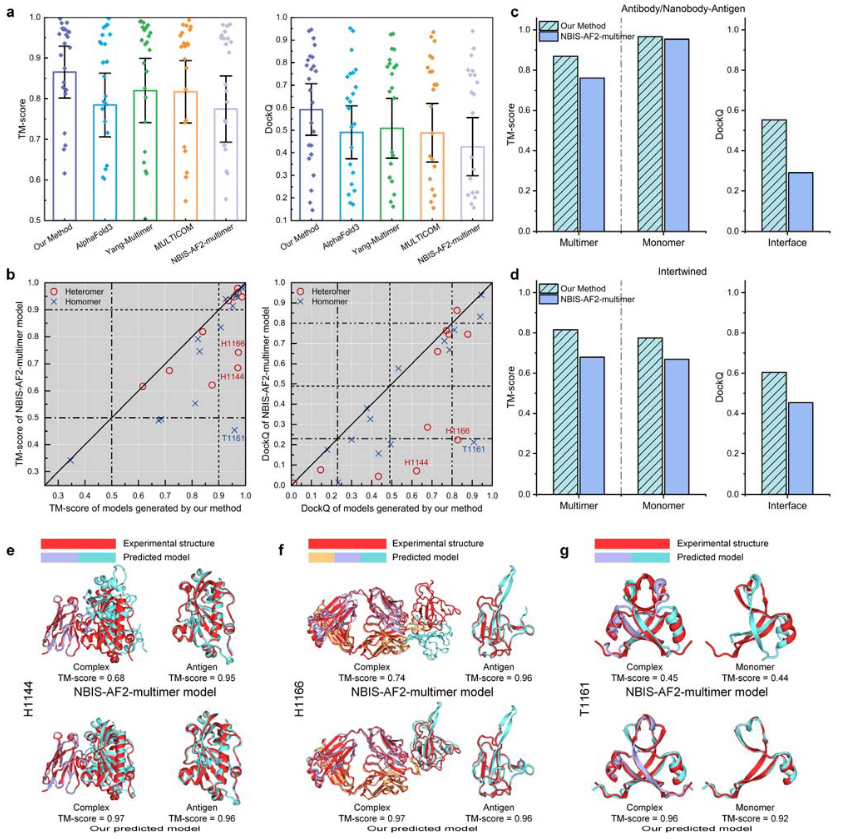
图2 DeepSCFold在CASP15数据集上蛋白质复合物结构预测的性能
DeepSCFold在CASP15多聚体目标基准集上的测试表现优异,显著超越了AlphaFold3、Yang-Multimer、MULTICOM和NBIS-AF2-multimer等现有方法。其平均TM-score达到0.87,DockQ分数为0.59,远高于其他方法(如AlphaFold3的TM-score为0.78和DockQ为0.49)。通过图2a可以看出,DeepSCFold在TM-score和DockQ方面均表现出色。
进一步分析显示,为评估DeepSCFold的增强输入策略在提高多聚体结构预测准确性方面的有效性,DeepSCFold与NBIS-AF2-multimer的直接比较,结果表明DeepSCFold在92%的案例中优于NBIS-AF2-multimer,TM-score提升显著;在88%的案例中DockQ分数更高(图2b)。特别是在复杂系统中,如缠绕复合物(T1161)、纳米抗体-抗原复合物(H1144)和抗体-抗原复合物(H1166),DeepSCFold的表现尤为突出。例如,在H1144中,尽管NBIS-AF2-multimer生成了高质量的单体模型(TM-score为0.95),但由于链间方向错误,复合物TM-score仅为0.68,而DeepSCFold通过更准确的链间信号将TM-score提升至0.97(图2e)。类似地,在H1166中,DeepSCFold将复合物TM-score从0.74提高到0.97(图2f)。
对于缠绕复合物(图2d),DeepSCFold同样展现了强大的性能,DeepSCFold在多聚体TM-score、单体TM-score和界面DockQ分数上均优于NBIS-AF2-multimer。以T1161为例,NBIS-AF2-multimer的单体TM-score仅为0.44,复合物TM-score低至0.45,而DeepSCFold分别将其提升至0.92和0.96(图2g)。这些结果表明,DeepSCFold通过整合序列衍生的结构互补性,不仅增强了链间距离和方向的确定性,还补充了传统共进化信号的不足,使复杂结构预测更加精准可靠。
此外,DeepSCFold在抗体-抗原复合物建模中的优势也十分明显(图2c)。其界面DockQ分数高达0.55,远超NBIS-AF2-multimer的0.29,显著提升了多聚体TM-score(NBIS-AF2-multimer为0.76,DeepSCFold为0.87)。
3.2 抗体-抗原复合物的DeepSCFold结构建模
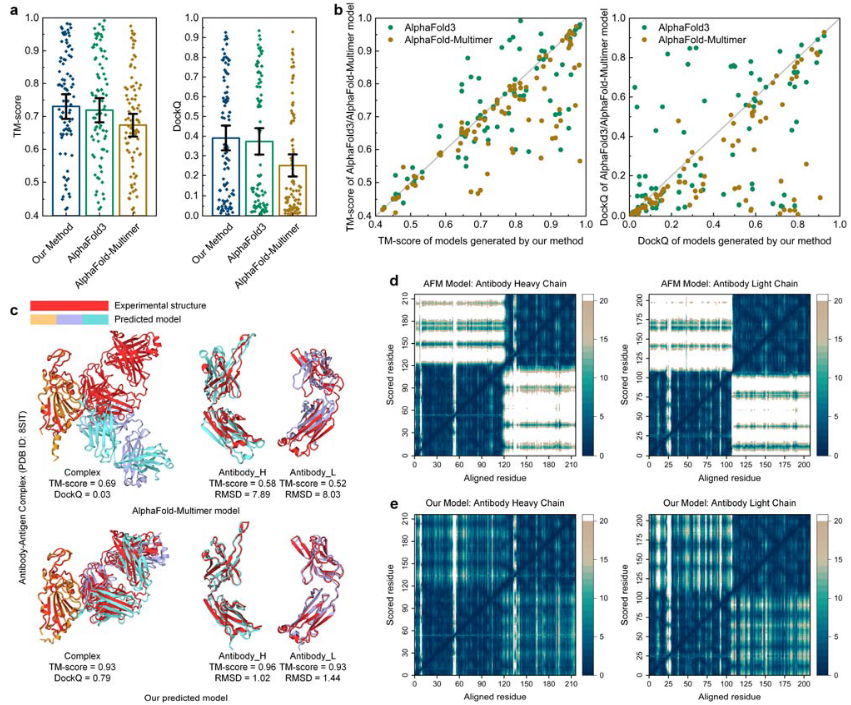
图3 抗体-抗原复合物结构预测的结果
抗体-抗原复合物因其缺乏显著的共进化信号,一直是蛋白质复合物建模中的难点。DeepSCFold通过创新的配对多序列比对(MSAs)和深度学习框架,展示了在抗体-抗原复合物结构预测显著优势(图3)。
DeepSCFold与AlphaFold-Multimer和AlphaFold3进行了对比测试,结果显示其在平均复杂TM-score(0.73)和界面DockQ(0.39)上均优于AlphaFold-Multimer(TM-score=0.67,DockQ=0.25)和AlphaFold3(TM-score=0.72,DockQ=0.37),表明其在整体复合物结构和界面质量预测上的优越性(图3a)。
以SARS-CoV-2 Spike RBD与抗体CC84.24 Fab复合物(PDB ID:8SIT)为例,AlphaFold-Multimer的预测模型在链间方向上存在显著误差,整体TM-score仅为0.63,界面DockQ低至0.03,且单体重链和轻链的TM-score分别为0.58和0.52,偏差较大(图3c、d)。而DeepSCFold不仅准确模拟了每个域的结构,还成功捕捉了域间和链间的取向,重链和轻链的PAE值在整个区域中均保持较低水平(图3e),显著提升了预测精度。
此外,在59.6%的测试案例中,DeepSCFold的TM-score高于AlphaFold3,在60.7%的案例中DockQ分数更高,并将抗体-抗原结合界面的预测成功率(DockQ>0.23)提升了12.4%。对于AlphaFold-Multimer无法准确建模的挑战性案例(DockQ<0.23),DeepSCFold成功构建了高质量模型,占比达39.3%(图3b)。这些结果表明,DeepSCFold基于结构互补性的配对MSAs为复杂建模提供了关键的链间相互作用信号,从而提高了复杂建模的准确性。
3.3 基于序列的结构相似度预测性能
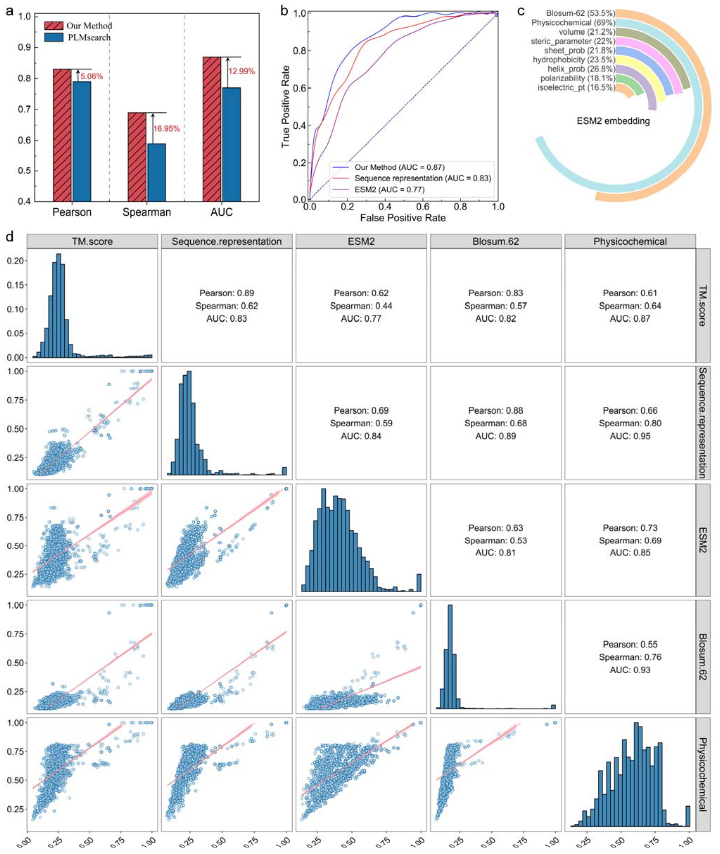
图4 蛋白质-蛋白质结构相似性预测的比较性能和特征分析
DeepSCFold实现了从序列数据直接预测蛋白质-蛋白质结构相似性的能力(图4)。
在基准测试中,DeepSCFold的表现远超现有的PLMsearch方法。图4a显示,DeepSCFold的皮尔逊相关系数达到0.83(比PLMsearch高5.06%),斯皮尔曼相关系数为0.69(高出16.95%),ROC AUC值为0.87(提升12.99%)。这些结果表明,DeepSCFold不仅在预测准确性上更优,还能更好地保留结构相似性的相对顺序,尤其适用于涉及蛋白质相似性排序的应用场景。
进一步分析表明,DeepSCFold的性能提升得益于对多样化特征的有效整合。图4b对比了ESM2嵌入与DeepSCFold生成的序列表示,结果显示单独使用ESM2的AUC值为0.77,而结合额外特征后提升至0.83,完整方法更是达到0.87。这凸显了分类模块的重要性,它能够充分利用组合特征实现更可靠的分类和排名性能。
图4c展示了ESM2嵌入与其他特征的相关性分析。整体理化性质特征集与ESM2嵌入的相关性最高(69.0%),表明两者捕获的生物学信息存在一定的重叠。个别特征如疏水性(23.5%)、螺旋概率(26.8%)和极化性(18.1%)的相关性相对较低,表明每个特征都提供了ESM2模型未能完全捕获的独特信息。Blosum-62与ESM2嵌入的相关性为53.5%,表明由替换概率编码的进化信息为结构相似性提供了互补的见解。
最后,图4d揭示了不同特征与真实TM分数的关系。序列表示与TM分数的相关性最高(Pearson’s r=0.89, Spearman’s ρ=0.62),表明整合多样化特征能有效捕捉全面的生物学信息。相比之下,单独的ESM2嵌入相关性较低(Pearson’s r=0.62, Spearman’s ρ=0.44),说明额外特征的加入显著提升了模型的预测能力。Blosum-62矩阵与TM分数的皮尔逊相关系数为0.83,斯皮尔曼相关系数为0.57,表明进化信息对结构相似性的贡献。
3.4 从序列上表现蛋白质-蛋白质相互作用概率
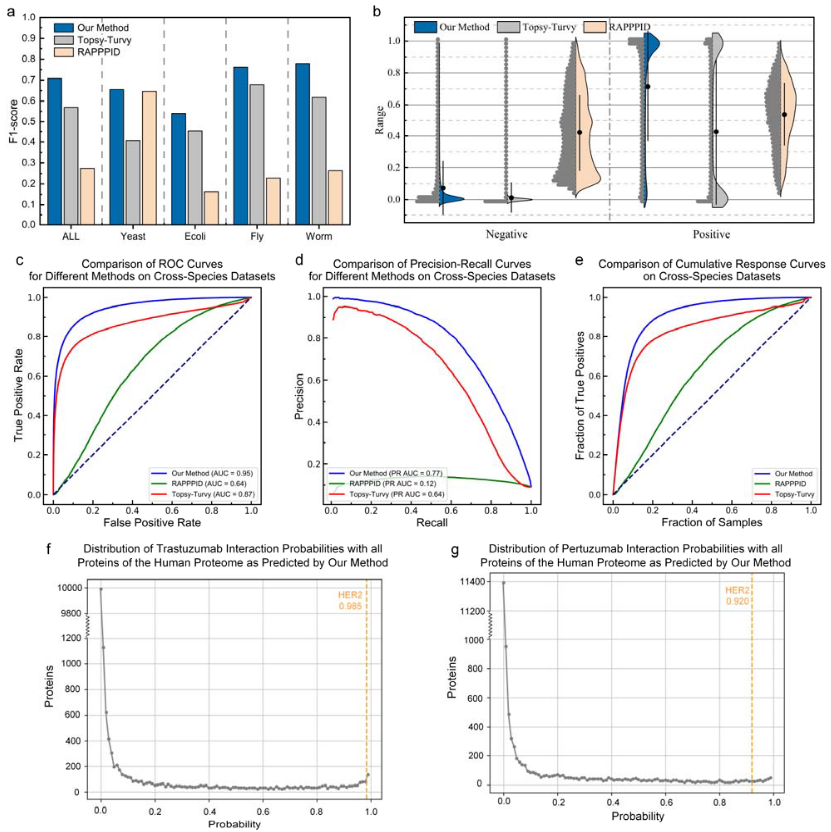
图5 蛋白质-蛋白质相互作用预测的比较分析与实际应用
DeepSCFold通过直接从序列数据预测蛋白质-蛋白质相互作用(PPI)概率,展现了其在复杂生物系统中的强大能力(图5)。与传统方法依赖实验数据和物种注释不同,DeepSCFold能够利用大规模未注释的蛋白质数据库(如BFD和MGnify),显著扩展了MSA构建的数据来源。
性能评估显示,DeepSCFold在四种模式生物(酵母、大肠杆菌、果蝇、线虫)及汇总数据集上的F1分数均优于Topsy-Turvy和RAPPPID(图5a),特别是在果蝇和线虫等具有复杂PPI网络的物种中表现尤为突出。图5b进一步揭示了其对正负样本的区分能力:负样本的概率分布接近零,而正样本集中在0.8以上,展现出高置信度的预测能力。相比之下,Topsy-Turvy的分布存在明显双峰,RAPPPID则缺乏足够的区分度。
ROC曲线分析中,其AUC值高达0.95,远超Topsy-Turvy(0.87)和RAPPPID(0.64)(图5c);PR曲线分析中,DeepSCFold的PR AUC为0.77,显著优于其他方法,体现了其在不平衡数据集中保持高精确度和召回率的能力(图5d)。此外,累积响应曲线CRC(图5e)表明,DeepSCFold能以更少的样本快速富集高置信度的PPIs,加速潜在相互作用的发现。
实际应用中,DeepSCFold展示了其在药物发现中的潜力。通过对曲妥珠单抗和帕妥珠单抗与人类蛋白质组的相互作用分析(图5f、g),模型成功预测了它们与HER2受体的高相互作用概率(分别为0.985和0.920),与已知生物学证据高度一致。这些结果不仅验证了DeepSCFold的可靠性,还突显了其在大规模相互作用筛选中的价值,为药物研发和蛋白质功能研究提供了强有力的支持。
4.讨论
准确捕获链间取向并预测蛋白质复合物结构是一个重大挑战,而DeepSCFold通过创新的深度学习方法显著提升了这一领域的准确性。其核心贡献在于利用深度学习网络捕捉潜在的结构互补性,并构建高质量的复合物多序列比对(MSAs)。实验结果表明,DeepSCFold在CASP15数据集上的TM-score较AlphaFold-Multimer和AlphaFold3分别提升了11.6%和10.3%,在抗体-抗原复合物建模中更是将界面预测成功率(DockQ > 0.23)提升了24.7%和12.4%。
DeepSCFold的优势在于克服了传统方法的局限性。例如,基于物种注释的序列连接机制仅限于基因组序列,而DeepSCFold充分利用宏基因组数据库中的信息,为单体MSAs的连接提供了新途径。此外,尽管抗体-抗原复合物因缺乏共进化信号而极具挑战性,DeepSCFold通过结构感知的复合物MSA构建,成功捕捉了形状和物理化学互补性,弥补了这一不足。
然而,DeepSCFold仍面临一些挑战。例如,用户需提供复合物的化学计量信息,这可能限制其实用性;准确的模型质量评估(MQA)对于区分高精度预测与错误预测至关重要,尤其是在缺乏实验验证的情况下。未来,DeepSCFold的应用场景可进一步拓展至DNA、RNA及配体-蛋白质相互作用建模,为更广泛的生物分子系统研究提供支持。
如何学习AI大模型 ?
“最先掌握AI的人,将会比较晚掌握AI的人有竞争优势”。
这句话,放在计算机、互联网、移动互联网的开局时期,都是一样的道理。
我在一线互联网企业工作十余年里,指导过不少同行后辈。帮助很多人得到了学习和成长。
我意识到有很多经验和知识值得分享给大家,故此将并将重要的AI大模型资料包括AI大模型入门学习思维导图、精品AI大模型学习书籍手册、视频教程、实战学习等录播视频免费分享出来。【保证100%免费】🆓
CSDN粉丝独家福利
这份完整版的 AI 大模型学习资料已经上传CSDN,朋友们如果需要可以扫描下方二维码&点击下方CSDN官方认证链接免费领取 【保证100%免费】

读者福利: 👉👉CSDN大礼包:《最新AI大模型学习资源包》免费分享 👈👈
对于0基础小白入门:
如果你是零基础小白,想快速入门大模型是可以考虑的。
一方面是学习时间相对较短,学习内容更全面更集中。
二方面是可以根据这些资料规划好学习计划和方向。
👉1.大模型入门学习思维导图👈
要学习一门新的技术,作为新手一定要先学习成长路线图,方向不对,努力白费。
对于从来没有接触过AI大模型的同学,我们帮你准备了详细的学习成长路线图&学习规划。可以说是最科学最系统的学习路线,大家跟着这个大的方向学习准没问题。(全套教程文末领取哈)

👉2.AGI大模型配套视频👈
很多朋友都不喜欢晦涩的文字,我也为大家准备了视频教程,每个章节都是当前板块的精华浓缩。


👉3.大模型实际应用报告合集👈
这套包含640份报告的合集,涵盖了AI大模型的理论研究、技术实现、行业应用等多个方面。无论您是科研人员、工程师,还是对AI大模型感兴趣的爱好者,这套报告合集都将为您提供宝贵的信息和启示。(全套教程文末领取哈)

👉4.大模型落地应用案例PPT👈
光学理论是没用的,要学会跟着一起做,要动手实操,才能将自己的所学运用到实际当中去,这时候可以搞点实战案例来学习。(全套教程文末领取哈)

👉5.大模型经典学习电子书👈
随着人工智能技术的飞速发展,AI大模型已经成为了当今科技领域的一大热点。这些大型预训练模型,如GPT-3、BERT、XLNet等,以其强大的语言理解和生成能力,正在改变我们对人工智能的认识。 那以下这些PDF籍就是非常不错的学习资源。(全套教程文末领取哈)

👉6.大模型面试题&答案👈
截至目前大模型已经超过200个,在大模型纵横的时代,不仅大模型技术越来越卷,就连大模型相关的岗位和面试也开始越来越卷了。为了让大家更容易上车大模型算法赛道,我总结了大模型常考的面试题。(全套教程文末领取哈)

👉学会后的收获:👈
• 基于大模型全栈工程实现(前端、后端、产品经理、设计、数据分析等),通过这门课可获得不同能力;
• 能够利用大模型解决相关实际项目需求: 大数据时代,越来越多的企业和机构需要处理海量数据,利用大模型技术可以更好地处理这些数据,提高数据分析和决策的准确性。因此,掌握大模型应用开发技能,可以让程序员更好地应对实际项目需求;
• 基于大模型和企业数据AI应用开发,实现大模型理论、掌握GPU算力、硬件、LangChain开发框架和项目实战技能, 学会Fine-tuning垂直训练大模型(数据准备、数据蒸馏、大模型部署)一站式掌握;
• 能够完成时下热门大模型垂直领域模型训练能力,提高程序员的编码能力: 大模型应用开发需要掌握机器学习算法、深度学习
CSDN粉丝独家福利
这份完整版的 AI 大模型学习资料已经上传CSDN,朋友们如果需要可以扫描下方二维码&点击下方CSDN官方认证链接免费领取 【保证100%免费】
读者福利: 👉👉CSDN大礼包:《最新AI大模型学习资源包》免费分享 👈👈

















 1084
1084

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









