









 这篇博客介绍了R语言中的PLS-DA(偏最小二乘判别分析)方法,用于处理分类和判别问题,特别是微生物组学数据。PLS-DA通过偏最小二乘法对数据进行旋转,以区分不同组间的样本。它是一种有监督的分析方法,对比了与无监督的PCA分析的区别,并详细阐述了`mixOmics`包中的`plsda`函数用法。此外,还提供了示例代码展示如何进行多重组别PLS-DA分析,并解释了如何解读分析结果,如VIP值等。
这篇博客介绍了R语言中的PLS-DA(偏最小二乘判别分析)方法,用于处理分类和判别问题,特别是微生物组学数据。PLS-DA通过偏最小二乘法对数据进行旋转,以区分不同组间的样本。它是一种有监督的分析方法,对比了与无监督的PCA分析的区别,并详细阐述了`mixOmics`包中的`plsda`函数用法。此外,还提供了示例代码展示如何进行多重组别PLS-DA分析,并解释了如何解读分析结果,如VIP值等。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
#The following initializes usage of Bioc devel
BiocManager::install(version='devel')
BiocManager::install("ropls")
- PLS-DA(Partial Least Squares Discriminant Analysis),即
偏最小二乘法判别分析,是多变量数据分析技术中的判别分析法,经常用来处理分类和判别问题。通过对主成分适当的旋转,PLS-DA可以有效的对组间观察值进行区分,并且能够找到导致组间区别的影响变量。
PLS-DA采用了经典的偏最小二乘回归模型,其响应变量是一组反应统计单元间类别关系的分类信息,是一种有监督的判别分析方法。因无监督的分析方法(PCA)对所有样本不加以区分,即每个样本对模型有着同样的贡献,因此,当样本的组间差异较大,而组内差异较小时,无监督分析方法可以明显区分组间差异;而当样本的组间差异不明晰,而组内差异较大时,无监督分析方法难以发现和区分组间差异。另外,如果组间的差异较小,各组的样本量相差较大,样本量大的那组将会主导模型。有监督的分析(PLS-DA)能够很好的解决无监督分析中遇到的这些问题。
与PCA分析的原理相同,PLS利用偏最小二乘法对数据结构进行投影分析。但PLS与PCA数据有本质的不同,PCA分析方法中只有一个数据集X,所有分析都只是基于这个的数据集,对应于一个多维空间。而PLS分析是建立在两个数据集X和Y基础上的,因此也就对应地存在两个多维空间,在利用投影方法计算PLS个主成分后,分别得到X和Y空间的两条轴线以及各个样本点在X和Y空间周上的得分t1、u1。对X和Y数据的关联分析就是将所有样本在X和Y空间个主成分轴上的得分t1、u1分别作相关分析,可以表示为ui1 = ti1+ri1,i表示不同样本,ri1表示残差。对应的,经过第二个主成分计算可以得到t2、u2,有关系式ui2 = ti2+ri2 。
如果用t1 、t2作图,表示数据集X的PCA得分图,而如果用t1、u1作图就表示个主成分下数据集X与数据集Y相关性。与PCA的载荷图(变量分布散点图)相类似,PLS可以用权重方式对X、Y数据集中的变量进行相关联,找出变量之间的关系。
PLS-DA只需要一个数据集X,但在分析时必须对样本进行指定分组,这样分组后模型自动加上另外一个隐含的数据集Y,该数据集变量数等于组别数,赋值时把指定的那一组规定为1,其他所有值均为0。其他计算方法与上述PLS方法相同。这种模型计算的方法强行把各组分门别类,有利于发现组间的异同点。
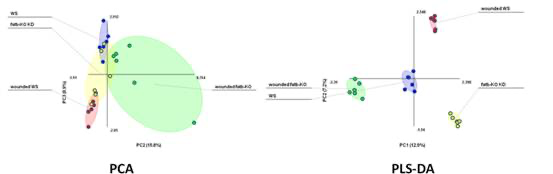
下图展示了无监督的PCA方法和有监督的PLS-DA之间的区别。
输入:
OTU Table文件:
OTU ID Bio1 Bio2 Bio3 Bio4 Bio5 Bio6 Bio7 Bio8 Bio9 Bio10
OTU1 0 0 0 0 0 6 34 104 367 254
OTU2 52 335 18 49 0 0 0 0 0 0
OTU3 0 0 0 0 5 0 0 0 0 0
样品分组信息表(可选):
Bio1 G1
Bio2 G1
Bio3 G1
Bio4 G1
Bio5 G2
Bio6 G2
Bio7 G2
Bio8 G3
Bio9 G3
Bio10 G3
输出:
plsda_sites.txt:记录了样本在各个维度上的位置,其中comp 1为x轴,comp 2为y轴。
plsda_rotation.txt:记录了OTU或者物种对各主成分的贡献度。
plsda_importance.txt:记录了各维度解释结果的百分比。如果plsda1值为50%,则表示x轴的差异可以解释全面分析结果的50%。
plsda_plot.pdf : plsda图
示例:Multiple groups plsda analysis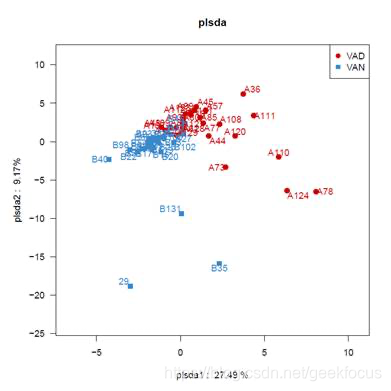
注:不同颜色或形状的点代表不同环境或条件下的样本组,横、纵坐标轴的刻度是相对距离,无实际意义。plsda1、plsda2分别代表对于两组样本微生物组成发生偏移的疑似影响因素,需要结合样本特征信息归纳总结,例如A组(红色)和B组(蓝色)样本在plsda1轴的方向上分离开来,则可分析为plsda1是导致A组和B组分开(可以是两个地点或酸碱不同)的主要因素,同时验证了这个因素有较高的可能性影响了样本的组成。
分析模块引用了R语言(v3.2.3)mixOmics包(v5.2.0)中的plsda分析和作图。
install.packages("mixOmics")
library(mixOmics)
plsda函数
Usage
plsda(X,Y,ncomp = 2,
scale = TRUE,mode = c("regression", "canonical", "invariant", "classic"),tol = 1e-06,max.iter = 100,near.zero.var = FALSE,
logratio="none", # one of "none", "CLR"
multilevel=NULL,all.outputs = TRUE)
Arguments
X numeric matrix of predictors. NAs are allowed.
Y a factor or a class vector for the discrete outcome.
ncomp the number of components to include in the model. Default to 2.
scale boleean. If scale = TRUE, each block is standardized to zero means and unit variances (default: TRUE)
mode character string. What type of algorithm to use, (partially) matching one of "regression", "canonical", "invariant" or "classic". See Details.
tol Convergence stopping value.
max.iter integer, the maximum number of iterations.
near.zero.var boolean, see the internal nearZeroVar function (should be set to TRUE in particular for data with many zero values). Setting this argument to FALSE (when appropriate) will speed up the computations. Default value is FALSE
logratio one of ('none','CLR') specifies the log ratio transformation to deal with compositional values that may arise from specific normalisation in sequencing dadta. Default to 'none'
multilevel sample information for multilevel decomposition for repeated measurements. A numeric matrix or data frame indicating the repeated measures on each individual, i.e. the individuals ID. See examples in ?splsda.
all.outputs boolean. Computation can be faster when some specific (and non-essential) outputs are not calculated. Default = TRUE.
data(breast.tumors)
X <- breast.tumors$gene.exp
Y <- breast.tumors$sample$treatment
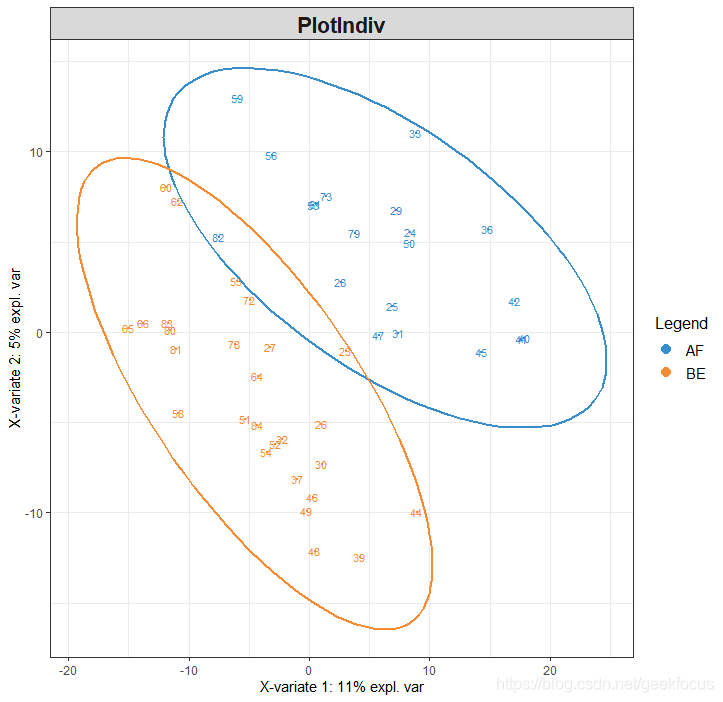
plsda.breast <- plsda(X, Y, ncomp = 2)
plotIndiv(plsda.breast, ind.names = TRUE, ellipse = TRUE, legend = TRUE)
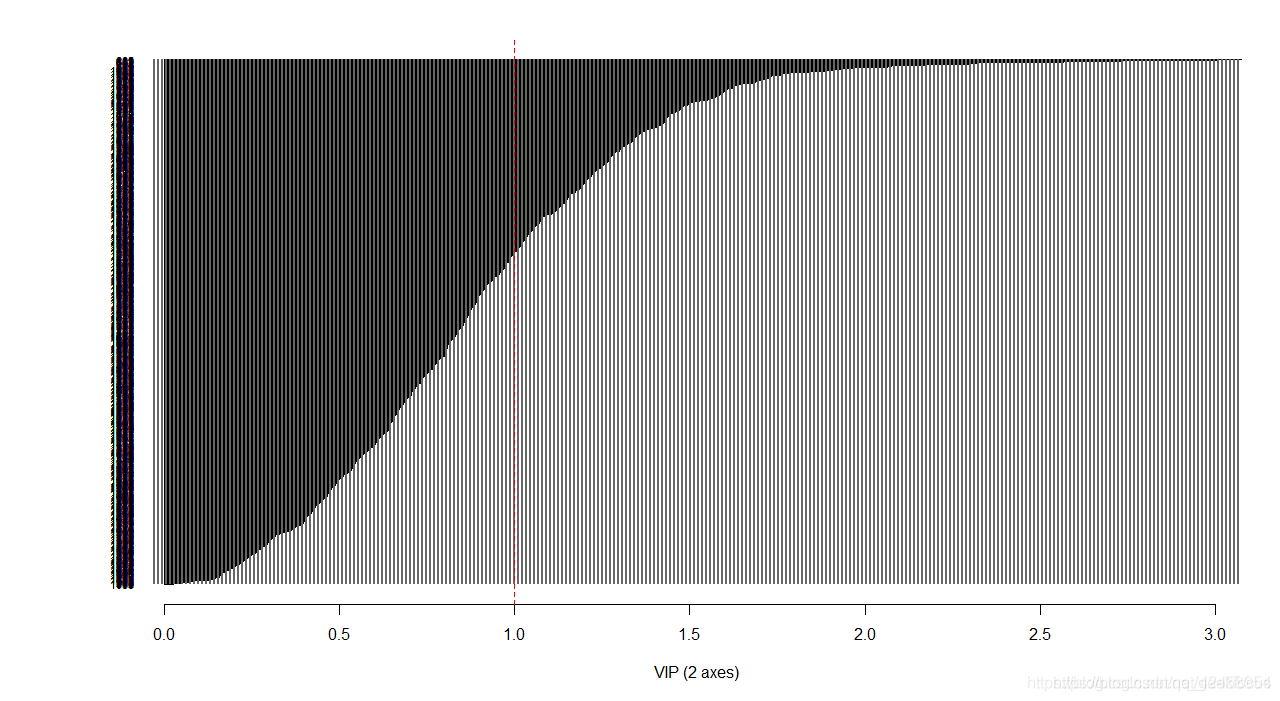
得到VIP值
library(RVAideMemoire)
PLSDA.VIP(plsda.breast, graph = TRUE)


















 1040
1040

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









