






Chemprop 是一个用于化学性质预测的机器学习软件包,其原理基于图神经网络,特别是有向消息传递神经网络(D-MPNN)架构。以下是其原理和具体过程:
1. 模型结构
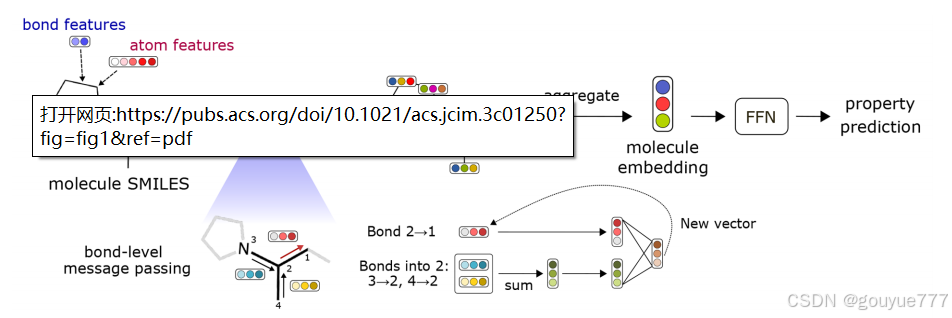
- 模块 1:局部特征编码
- 输入分子的 SMILES 字符串,通过 RDKit 转换为分子图,原子对应顶点,键对应边。
- 为每个顶点(原子)和边(键)构建初始特征向量。原子特征包括原子序数、键数、形式电荷、手性、氢原子数、杂化方式、芳香性和原子质量等信息的独热编码;键特征包括键类型、是否共轭、是否在环中以及是否包含立体化学信息。
- 还可读取自定义的原子和键特征。
- 模块 2:有向消息传递神经网络(D-MPNN)
- 消息在有向边之间传递,而非传统的节点之间。
- 首先通过单个神经网络层和非线性激活函数(默认 ReLU)将初始有向边特征转换为隐藏有向边特征,用户可选择激活函数和隐藏特征大小。
- 然后基于局部环境通过 T(默认 3)个消息传递步骤迭代更新有向边特征,提高数值稳定性会排除反向的有向边。
- 最后将更新后的隐藏状态聚合成原子嵌入,用户可定制 D-MPNN 的超参数,如激活函数、隐藏大小和消息传递步骤等。
- 模块 3:聚合函数
- 将分子中所有原子的嵌入聚合成单个分子嵌入。
- 提供三种聚合选项:求和、缩放求和(除以用户指定的缩放因子)和平均(默认)。
- 可选择提供额外的分子特征向量,该向量可以是外部来源提供的特征或生成的工程指纹(如 Morgan 圆形指纹和 RDKIT 2D 指纹)。
- 模块 4:标准前馈神经网络(FFN)
- 从分子嵌入中学习分子目标性质,用户可选择层数(默认 2 层)和隐藏神经元数量(默认 300 个)。
- 激活函数与 D-MPNN 相同,默认开启偏置。对于二元分类任务,最终模型输出通过 sigmoid 函数约束在(0,1)范围内;对于多分类任务,使用 softmax 函数使分类分数在各类别上总和为 1。
2. 训练过程
- Chemprop 是完全端到端可训练的,D-MPNN 和 FFN 的权重同时更新。
- 默认使用随机数据分割训练单个模型,共 30 个轮次,但小数据集可能需要更多轮次,建议检查学习曲线的收敛性。
- 使用 Adam 优化器,默认学习率在前两个热身轮次从线性增加到,之后在剩余轮次从指数下降到,每个优化步骤默认使用大小为 50 的数据点批次。
- 提供早期停止和丢弃(dropout)作为正则化手段,其 PyTorch 后端支持 GPU 加速训练和推理过程。
3. 特色功能
- 支持多种输入特征
- 除了 SMILES 字符串外,还可接受分子、原子或键级别的额外特征作为输入,外部信息作为附加特征可进一步提高性能。
- 多分子模型
- 可以在包含多个分子的数据集上进行训练,如溶质 / 溶剂组合或有 / 无溶剂的反应相关性质预测。
- 默认对每个分子训练单独的 D-MPNN,也可指定使用相同的 D-MPNN,然后将不同分子的嵌入连接后再输入到 FFN。
- 反应支持
- 通过使用特定关键字支持原子映射反应的输入,将反应物和产物的 SMILES 对转换为一个伪分子(凝聚反应图,CGR),然后传递给常规的 D-MPNN 模块。
- 可选择接受额外的分子对象(如溶剂、试剂等)作为输入,其输出与反应 D-MPNN 的输出在原子聚合后、FFN 之前连接。
- 光谱数据支持
- 支持分子的全光谱性质预测,使用光谱信息散度(SID)作为损失函数,可处理数据集中有间隙或缺失值的目标,还能创建排除区域以忽略无效的光谱部分进行训练。
- 潜在表示
- 学习到的节点表示在消息传递阶段后聚合成分子级别的 “学习指纹”,在 FFN 网络中还有最终隐藏表示 “ffn 嵌入”,这些潜在表示可用于数据聚类或作为其他模型的附加特征。
- 损失函数选项
- 根据数据集类型(回归、分类、多分类或光谱)可使用多种常见损失函数进行模型训练,如均方误差(MSE)、有界 MSE、负对数似然(NLL)等,同时也提供一些不可微的度量用于模型评估。
- 迁移学习
- 可将从一个预训练模型中学习到的模型参数转移到新模型中,包括初始化新模型参数同时正常更新转移的权重,或者冻结转移模型的部分参数(如 D-MPNN 权重以及可指定的一些 FFN 层)。
- 超参数优化
- 提供命令行工具用于启动超参数优化任务,可指定试验次数和要搜索的超参数,先随机采样试验,然后使用基于树结构的 Parzen 估计器算法进行有针对性的采样,支持多实例并行操作。
- 不确定性工具
- 包含多种流行的不确定性估计、校准和评估工具,如深度集成、丢弃法、均方差估计等,还提供多种校准方法用于不同任务,并包含一些专门用于评估不确定性估计质量的指标。
- 原子 / 键级目标
- 支持多任务约束的 D-MPNN 架构用于预测原子和键级别的性质,如电荷密度或键长,可同时训练多个原子和键的性质,还可使用基于注意力的约束方法确保预测的原子或键性质总和符合指定的分子净值。


















 2001
2001

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









