









(一)量子化学计算如何选择硬件配置
由于分子的类型、体系和模拟算法,以及各种应用和精度要求,都有不同的计算特点,对计算机硬件(CPU、内存、硬盘、GPU)配置要求有很大的差异 ,那么如何选择好服务器/工作站的硬件配置,提高计算速度,缩短求解时间?主要看用量子化学计算软件的算法,及做什么求解,因为算法和精度决定了计算特点和求解规模,合理的硬件配置,大大发挥机器性能,缩短求解时间。
主要量子化学软件:Gaussian,GAMESS,MOLPRO,NWChem

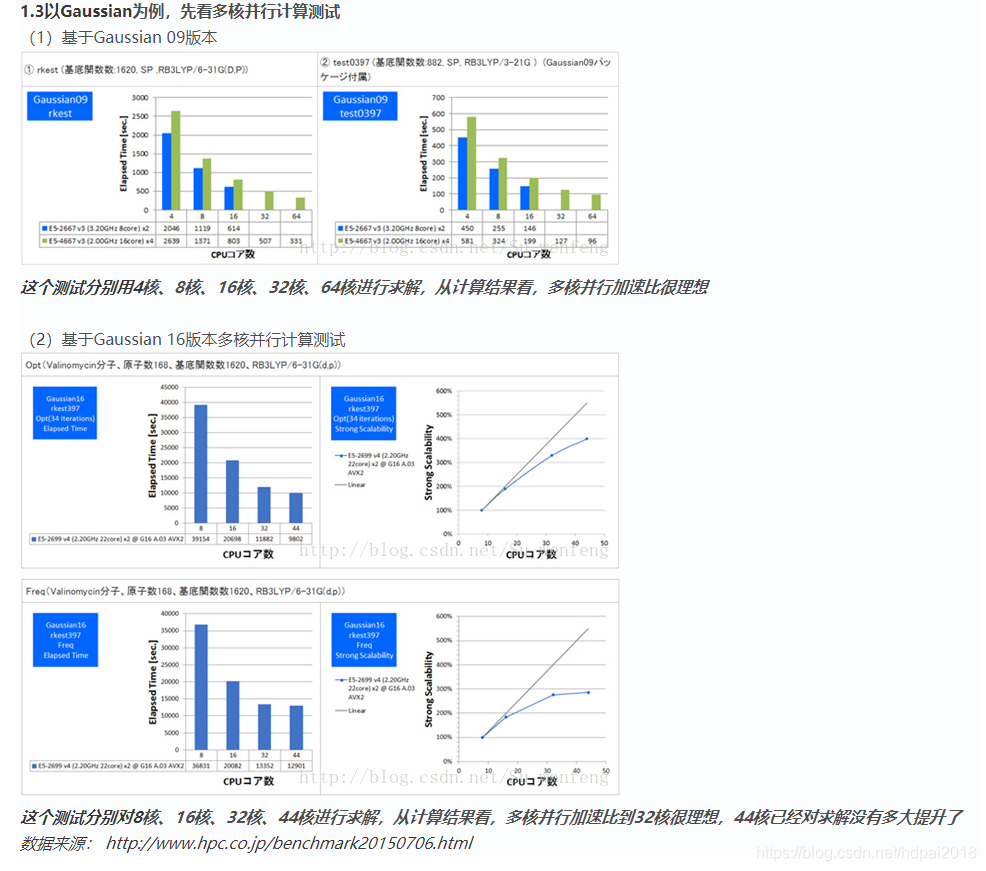
1.4 量子化学计算硬件配置特点
(1)不同算法的并行计算的核数加速比有所差异,不完全是线性的
(2)最新CPU架构和提升频率,对缩短求解时间非常显著
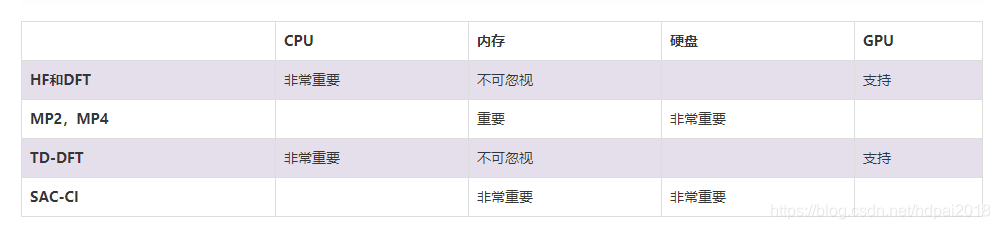
(3)精度高求解计算量大,对CPU频率和核数、内存容量、硬盘io要求均有很高要求
数据来源:http://www.hpc.co.jp/chem/c_select_hardware.html
(4)核数与内存比例为 1:4最为理想
(5)支持Tesla K40或K80 的GPU计算,显存需12GB以上,但算法有局限性,同时性价比不好
(6)组成集群的计算节点配置最好采用4核~10核超高频,效率最高
(二)基于量子化学计算,UltraLAB完美硬件配置推荐
2.1 UltraLAB图形工作站机型介绍
UltraLAB是西安坤隆计算机科技有限公司推出的定制图形工作站品牌,经过多年发展,该产品拥有傲视群雄的三大领先优势:先进计算硬件架构、完整齐全行业应用定制方案、专业硬件系统优化技术,大幅超越同类的“图形工作站”产品。以下是基于量子化学计算应用最快硬件架构产品系列:

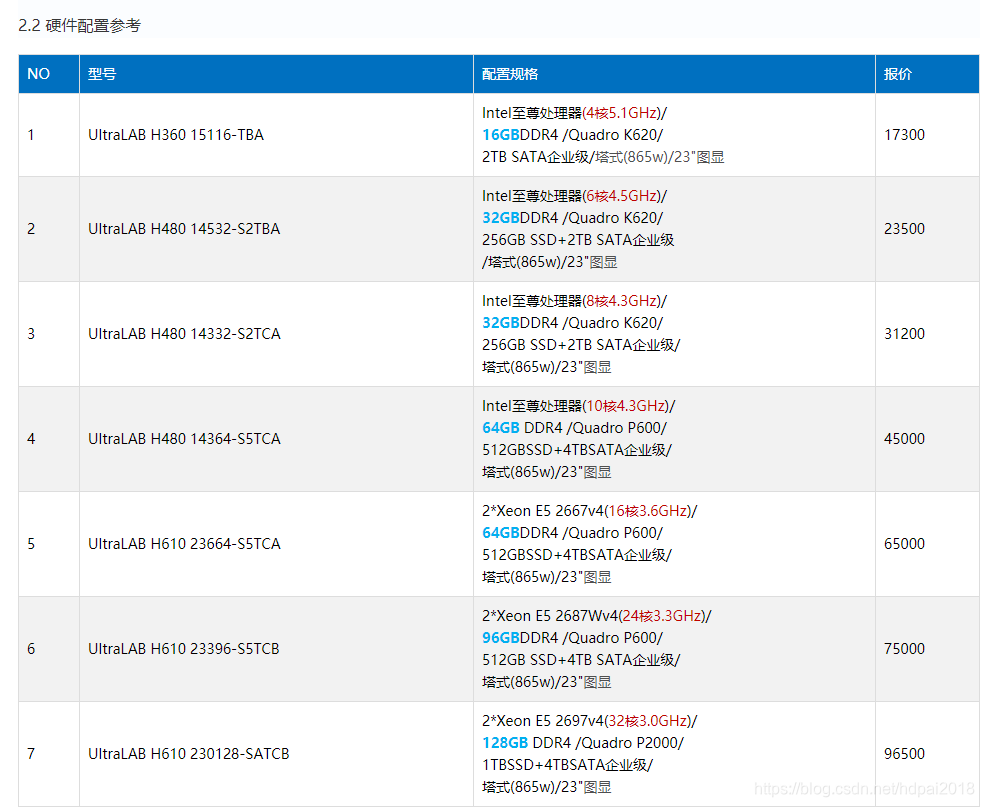
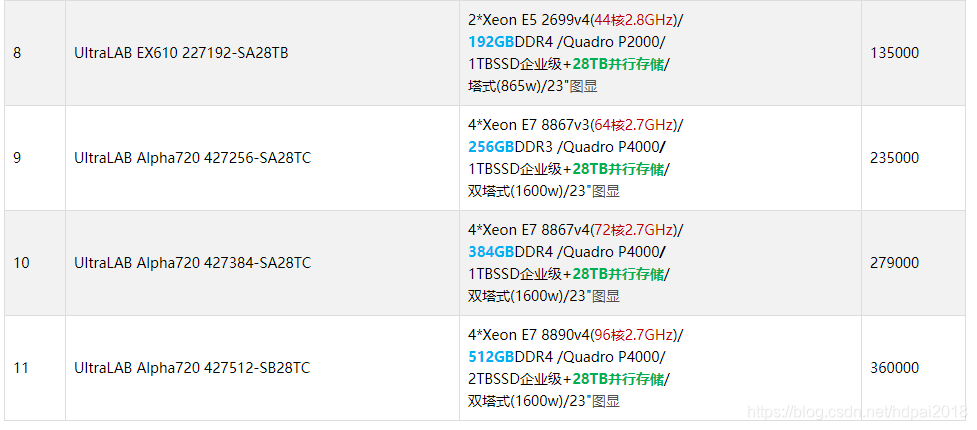
备注:以上价格仅供参考
上述配置其显著特点,
(1)全部采用最新intel硬件架构
(2)提供不用规模的高效完美硬件配置,满足不同算法和精度计算需求
(3)每个配置架构,CPU频率达到极致,提升性能关键指标
(4)足够多核数,并行存储高io,满足高精度计算,均衡无死角
(5)整机全部提供系统优化,大幅提升计算速度
(6)支持GPU计算扩展
————————————————
版权声明:本文为CSDN博主「於無聲處聽驚雷」的原创文章,遵循 CC 4.0 BY-SA 版权协议,转载请附上原文出处链接及本声明。
原文链接:https://blog.csdn.net/Su_wenfeng/article/details/79456396

















 1641
1641

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









