






愿武艺晴小朋友一定得每天都开心
从Fragements文件到和greenleaf比较相似的UMAP图形就长这样了。放过自己了,具体过程如下:
那就先去学习学习ArchR 理论知识(属于个人的理解分类和总结)
1)ArchR 在peak 注释(peak 注释不等于Peak calling,而是包含Peak calling)上采用Bin/window的方式;理解为:把基因组切割成不重叠的固定长度区间(通常是5kb);其中ArchR中Bin的长度默认为500bp得到TileMatrix矩阵,这种小的Bin能够更精确地检测到TF的结合位点或位置范围(300-500bp);我感觉:能做到500bp 的bin且对硬件需求大众化,运行也良好,得益于greenleaf团队采用的Arrow文件方法。
2)ArchR 采用两阶段的方法进行Peak Calling:1)先用bin长度(默认为500bp)得到的TileMatrix矩阵(Bin-cell)矩阵并作聚类,2)然后把得到的每一个类中所有的Read汇总起来(PeakMatrix)做Peak Calling.这里ArchR采用了MACS2的方法
接着上一篇;就要针对PeakMatrix进行参数的调整了:

从跟作者像的角度,我们发现varFeatures =6800 ,是比较合适的
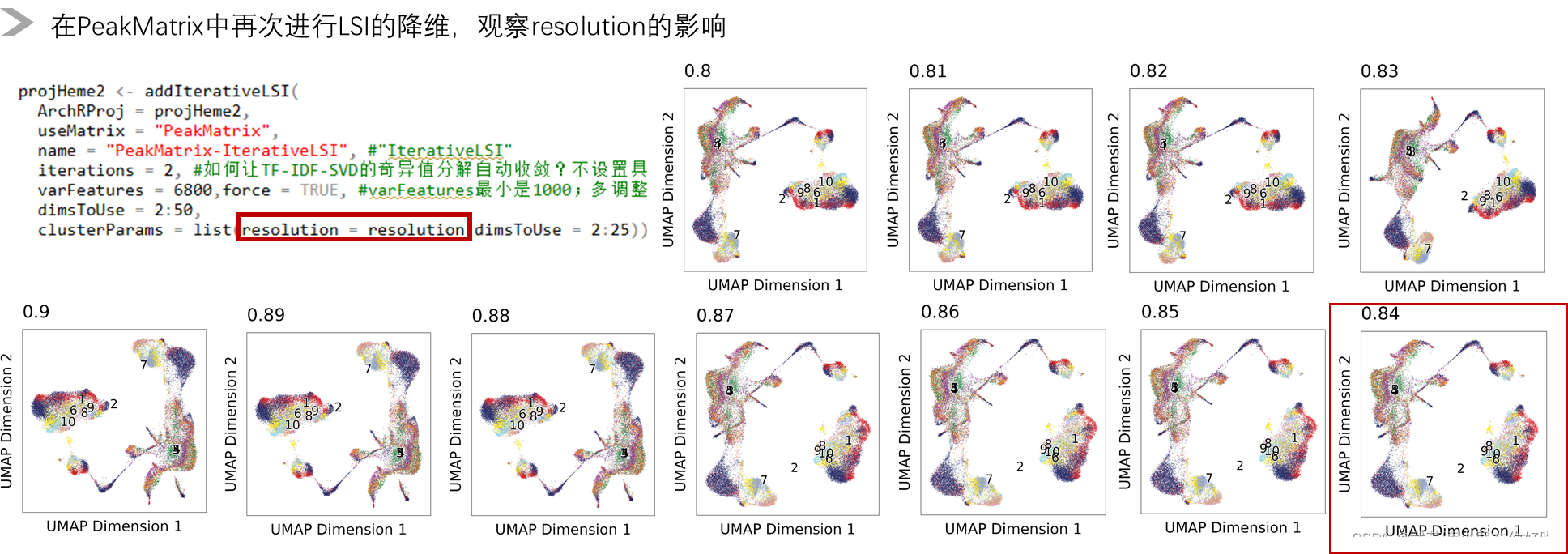
从跟作者像的角度,我们发现resolution =0.84/0.85/0.86/0.87都是比较合适的
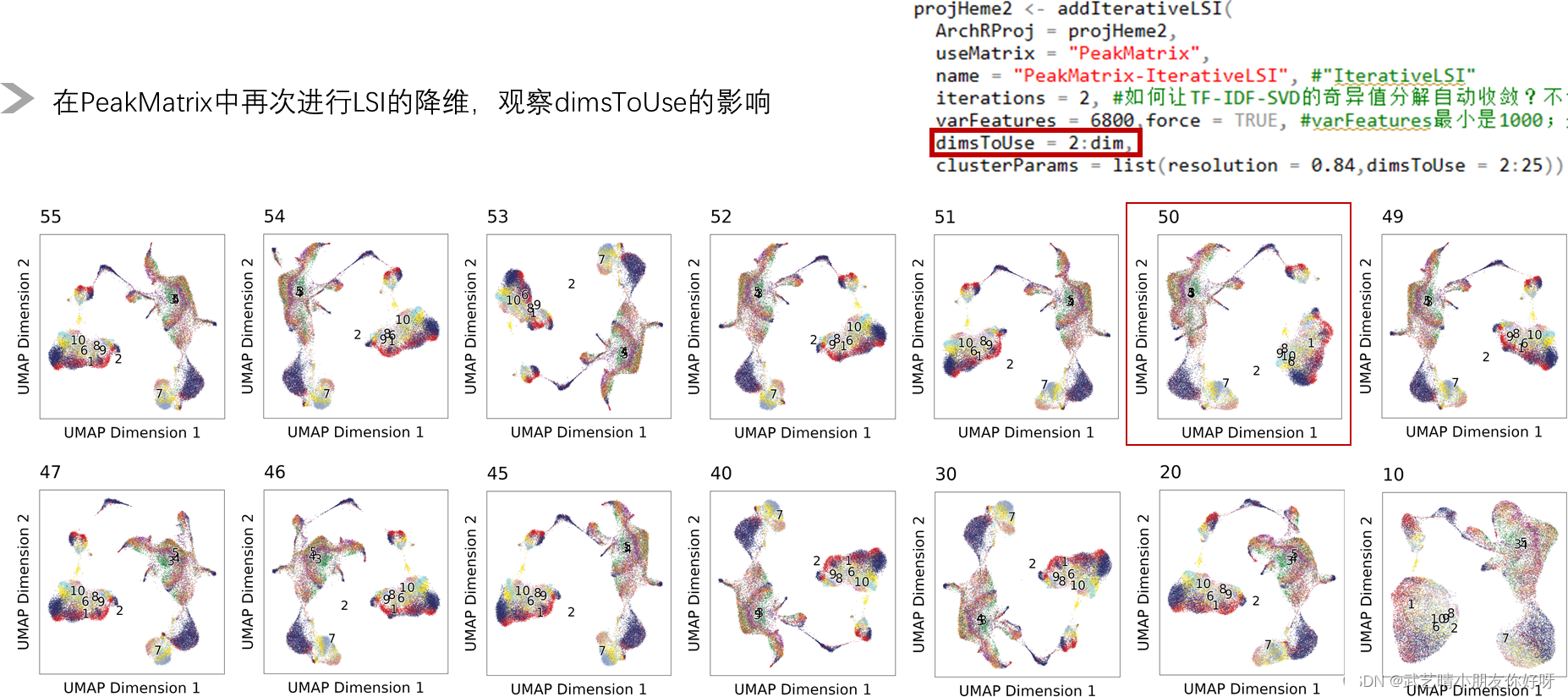
从跟作者一致的角度,我们发现dimsToUse =50 是比较合适的一个
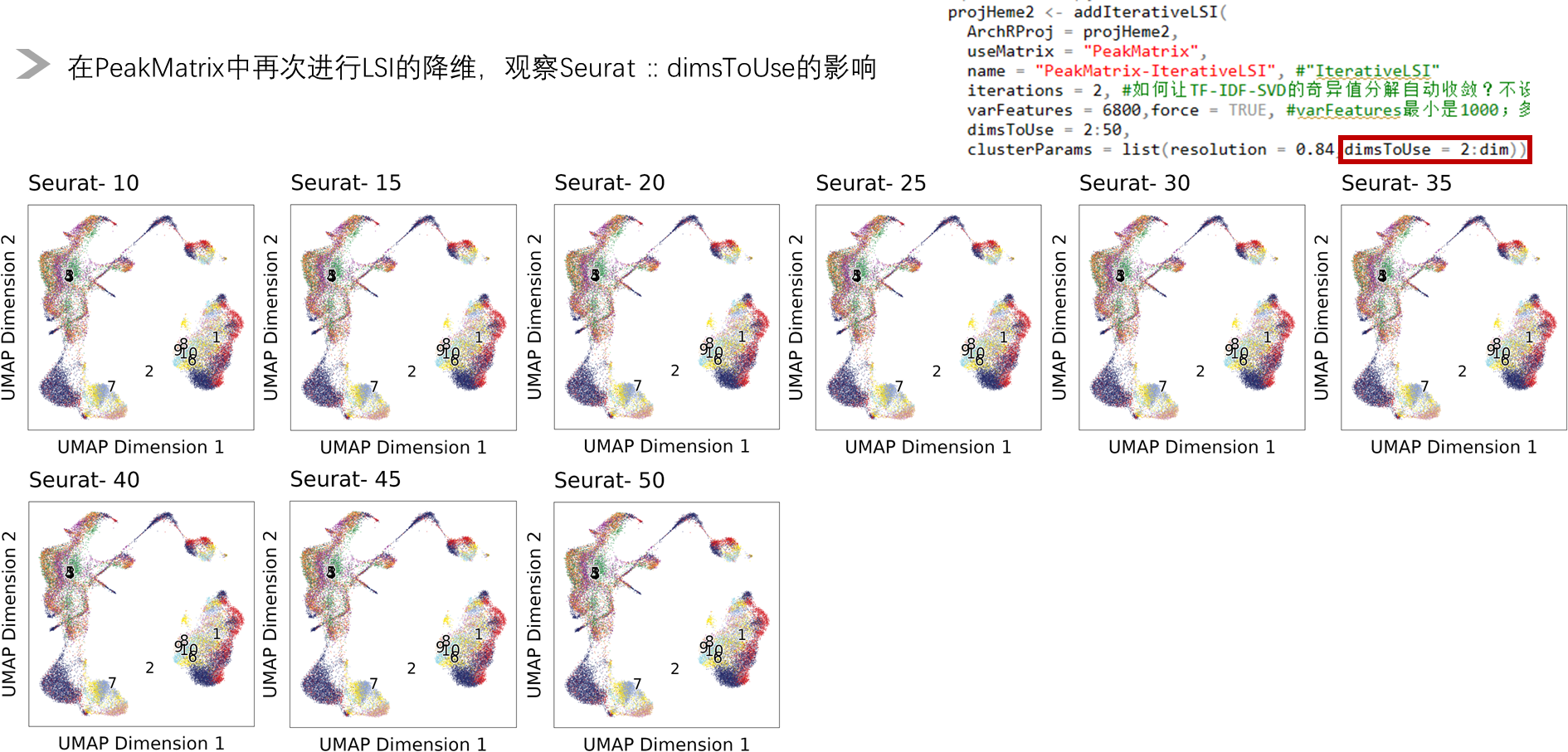
从跟作者一致的角度,我们发现Seurat::dimsToUse 对UMAP的降维无影响
最后综合以上的摸索,感觉还行吧,放过自己了:
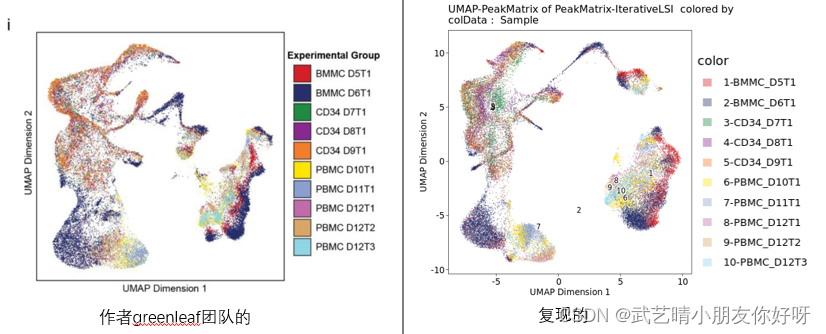
作者show出的Marker基因的情况:
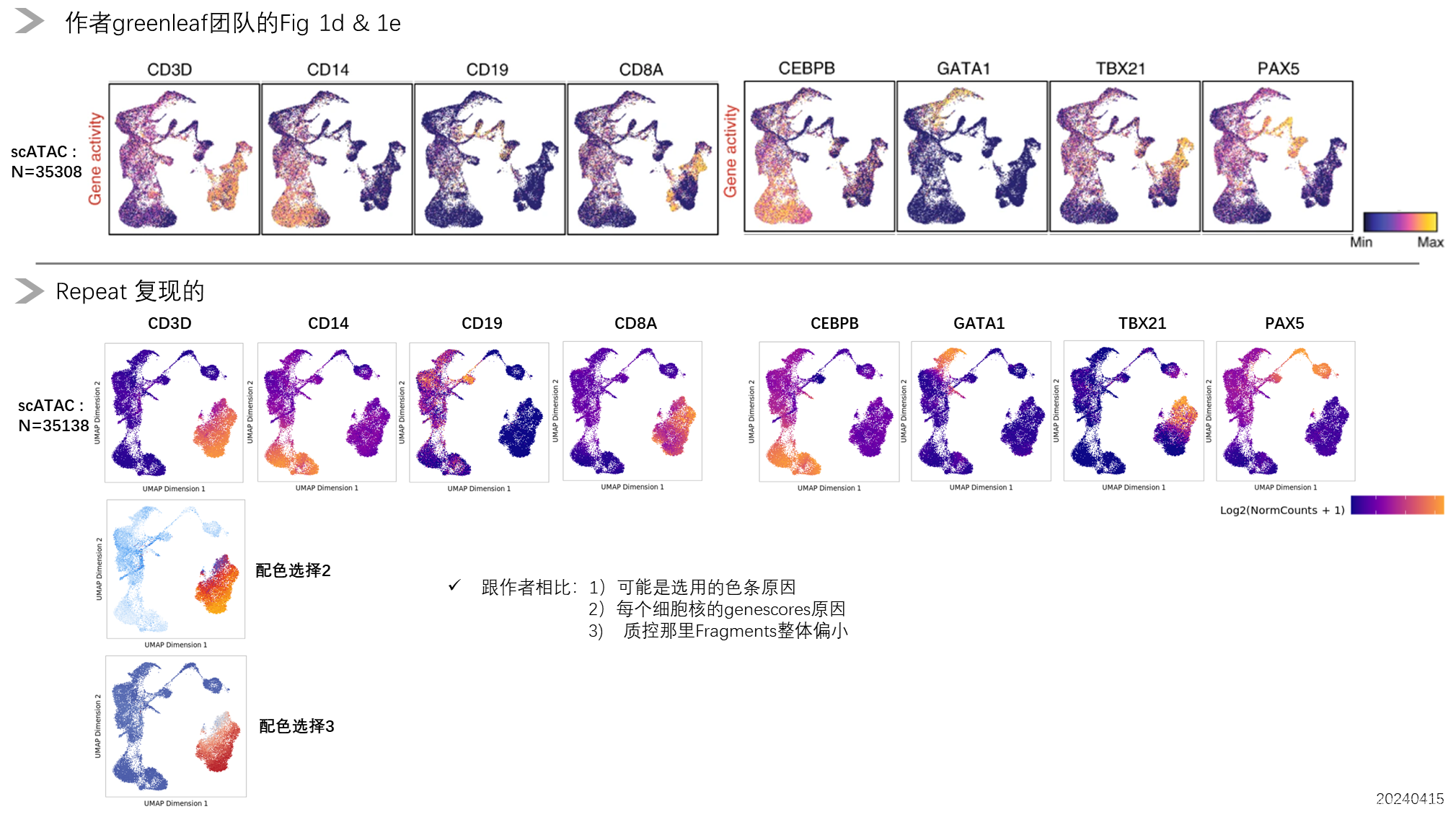
最后最后,恳请走过路过的朋友们留下您们宝贵的意见。争取一起努力把ArchR学会至精通。
我写博客的初衷是发现,scATAC-seq的中文资源和教程相比scRNA-seq少的可怜。既然我开始学了,为了以后大家少走些弯路,那就写把。毕竟网络是大家最好的老师,当是一起学习了。















 1664
1664

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









