






MicrobiomeAnalyst是一个方便易用的网页工具,是系统全面分析微生物组学数据的分析网站。
https://www.microbiomeanalyst.ca
所需的Data文件如下图所示。
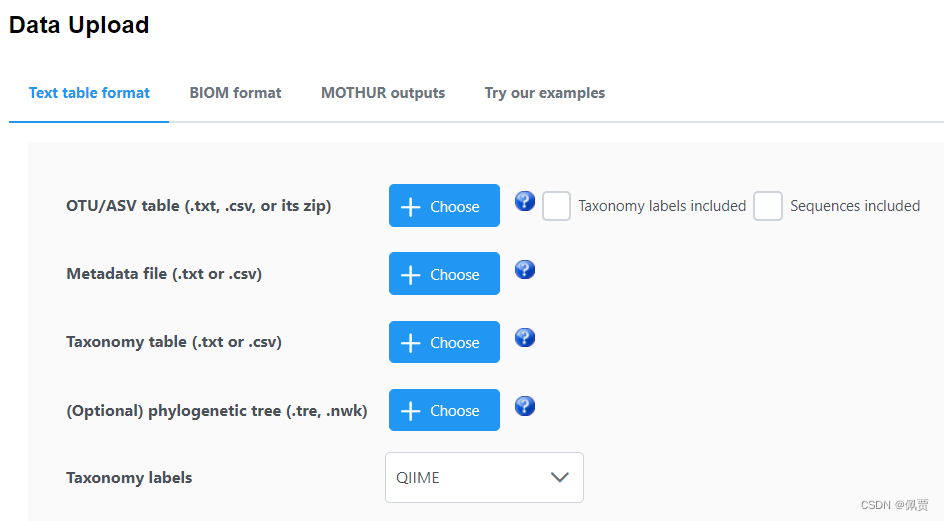
一、公司文件(以美吉为例)
1.OTU/ASV table(Taxonomy labels included)
文件位置:workflow_results\ASVTaxon_summary\asv_taxon.xls
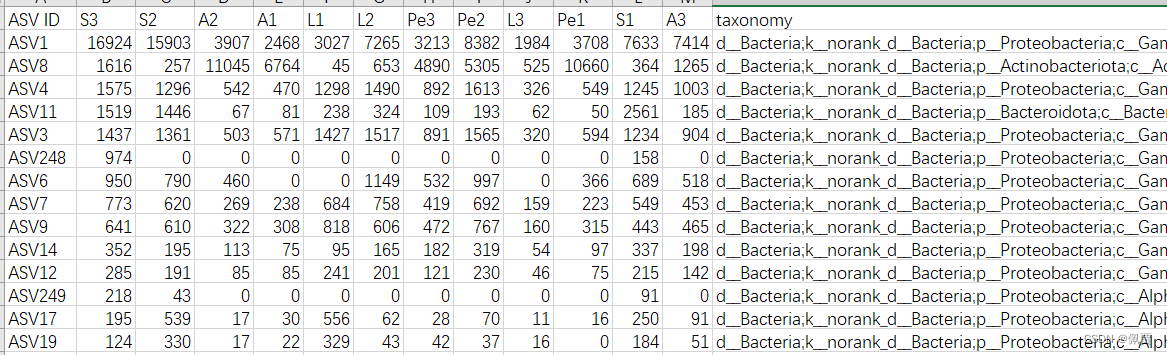
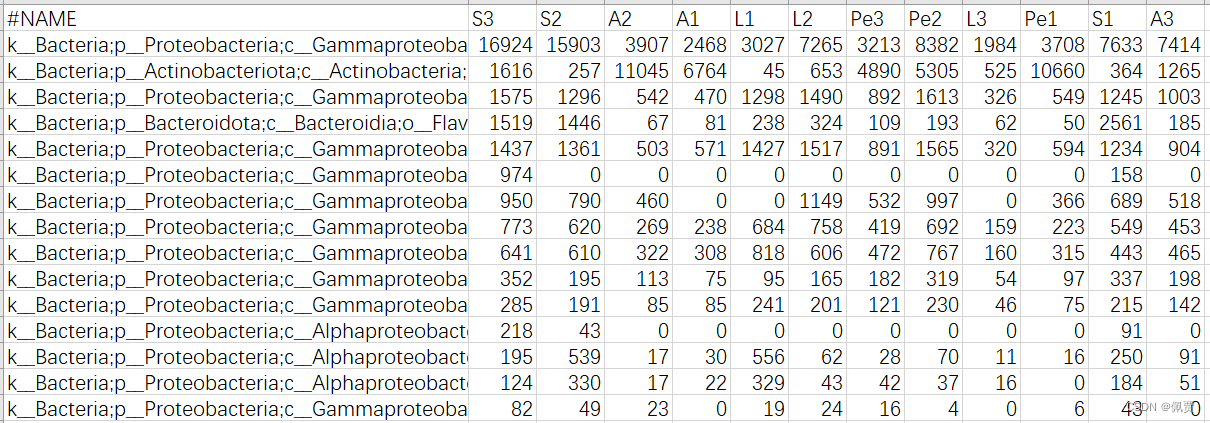
将ASV ID一列删除;将taxonomy一列移动到最前面(需要删除其中k__norank_d__Bacteria分类等级,将d__Bacteria改为k__Bacteria);将taxonomy改为#NAME
修改后
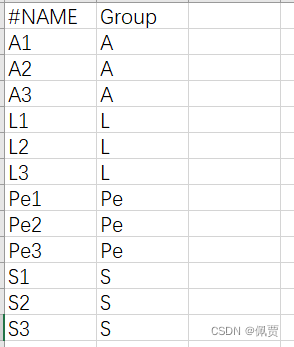
2.Metadata file
自行编辑
3.(Optional) phylogenetic tree
文件位置:workflow_results\ASV\ASV_phylo.tre
二、QIIME 2生成的文件
1.所需原始文件:

1.1 seq(样本序列):扩增子测序文件(gz格式或者fastq和fasta格式)

1.2 manifest(样本列表):第一列为样本的ID,第二列为每个样本测序文件的路径。
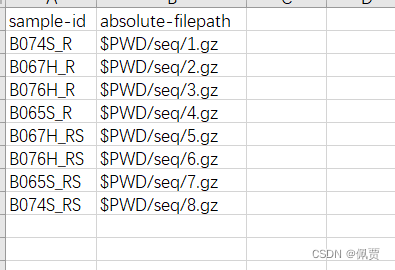
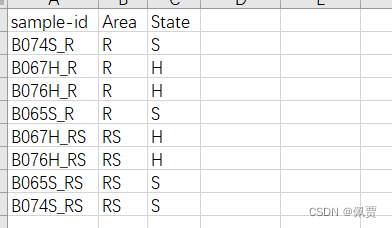
1.3 metadata.txt(Meta信息):第一列为样本的ID,其他列为meta信息(例如分组)
1.4 分类器(classifier)
# 训练分类器
# 下载GreenGenes并解压
wget http://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
tar -xzvf gg_13_8_otus.tar.gz
# 导入参考序列
qiime tools import \
--type 'FeatureData[Sequence]' \
--input-path gg_13_8_otus/rep_set/99_otus.fasta \
--output-path 99_otus.qza
# 导入物种分类信息
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \
--input-path gg_13_8_otus/taxonomy/99_otu_taxonomy.txt \
--output-path ref-taxonomy.qza
# 提取特定区段序列(引物序列可自行修改,此处为799F-1193R)
time qiime feature-classifier extract-reads \
--i-sequences 99_otus.qza \
--p-f-primer AACMGGATTAGATACCCKG \
--p-r-primer ACGTCATCCCCACCTTCC \
--o-reads ref-seqs_G_799F-1193R.qza
# 生成特定分类器
time qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads ref-seqs_G_799F-1193R.qza \
--i-reference-taxonomy ref-taxonomy.qza \
--o-classifier classifier_G_799F-1193R.qza2.QIIME 2处理:
2.1 文件生成
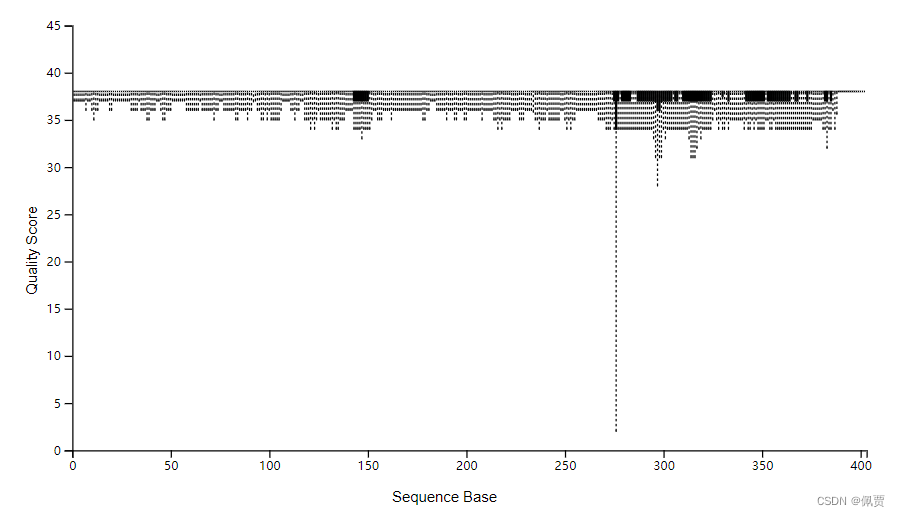
Quality Score中位数至少大于20,结果显示序列不用剪切。
# 创建data文件夹
mkdir data_MicrobiomeAnalyst
# 数据导入(质量值33类型的单端数据)
time qiime tools import \
--type 'SampleData[SequencesWithQuality]' \
--input-format SingleEndFastqManifestPhred33V2 \
--input-path manifest \
--output-path demux.qza
# 结果可视化(用于DADA2参数调整)
# Quality Score中位数至少大于20
time qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
# 生成特征表和代表序列(DADA2降噪)
# n-threads:线程数,0为使用全部线程
# trim-left:序列剪切起点,0为不剪切
# trunc-len:序列剪切终点,0为不剪切
time qiime dada2 denoise-single \
--i-demultiplexed-seqs demux.qza \
--p-n-threads 0 \
--p-trim-left 0 \
--p-trunc-len 0 \
--o-representative-sequences rep-seqs.qza \
--o-table table.qza \
--o-denoising-stats stats.qza
cp table.qza data_MicrobiomeAnalyst/
# 生成进化树
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
cp rooted-tree.qza data_MicrobiomeAnalyst/
# 生成物种注释文件
qiime feature-classifier classify-sklearn \
--i-classifier /mnt/e/bioinfo/storage/QIIME2/classifier/database/classifier_G_799F-1193R.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
cp taxonomy.qza data_MicrobiomeAnalyst/2.2 文件处理
# 进入data文件夹
cd data_MicrobiomeAnalyst
# 1.OTU/ASV table
# table.qza → table.rar → feature-table.biom
biom convert -i feature-table.biom -o table.txt --to-tsv
# feature-table.biom → table.txt
# 2.Metadata file
# 自行编辑
# 3.Taxonomy table
# taxonomy.qza → taxonomy.rar → taxonomy.tsv
# 4.(Optional) phylogenetic tree
# rooted-tree.qza → rooted-tree.rar → tree.nwk三、QIIME2处理公司文件
文件位置:
workflow_results\ASV\ASV_table.qza
workflow_results\ASV\ASV_reps.qza
# 创建data文件夹
mkdir data_MicrobiomeAnalyst
# 放入数据
cp ASV_table.qza data_MicrobiomeAnalyst/
# 生成进化树
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences ASV_reps.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
cp rooted-tree.qza data_MicrobiomeAnalyst/
# 生成物种注释文件
qiime feature-classifier classify-sklearn \
--i-classifier /mnt/e/bioinfo/storage/QIIME2/classifier/database/classifier_G_799F-1193R.qza \
--i-reads ASV_reps.qza \
--o-classification taxonomy.qza
cp taxonomy.qza data_MicrobiomeAnalyst/文件处理同上文中2.2
附:
1.安装Biological Observation Matrix (BIOM)
# 创建环境
conda create -n biom
# 进入环境
conda activate biom
# 安装numpy支持包
conda install numpy
# 安装biom
conda install -c conda-forge biom-format
# biom转换为txt
conda activate biom
biom convert -i feature-table.biom -o table.txt --to-tsv2.OTU/ASV table(Taxonomy labels included)不能生成Phylogenetic Tree Analysis

















 615
615

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









