










本文内容主要包括三个部分:
- 生物序列 tokenization 的原理讲解
- 手动实现一个简单的 Tokenizer
- 将 fasta 文件转变为预训练模型要求的数据输入格式:k-mer 格式
其中,第 2 和第 3 部分主要为代码实现讲解。
1. 原理讲解
tokenization是自然语言处理领域非常成熟的一项技术,tokenization就是把我们研究的语言转换成计算机能够识别的数字——token。
在生物领域,如何把核苷酸或氨基酸序列tokenization成token呢?
我们可以使用k-mer技术:
k-mer指的是将k个序列单元作为一个滑动窗口,从第一个序列单元开始扫描,提取出序列中的k-mer。然后我们再对k-mer建立索引。
如下图所示,我们采用了3-mer窗口将“AGCACT”每次滑动1个碱基。
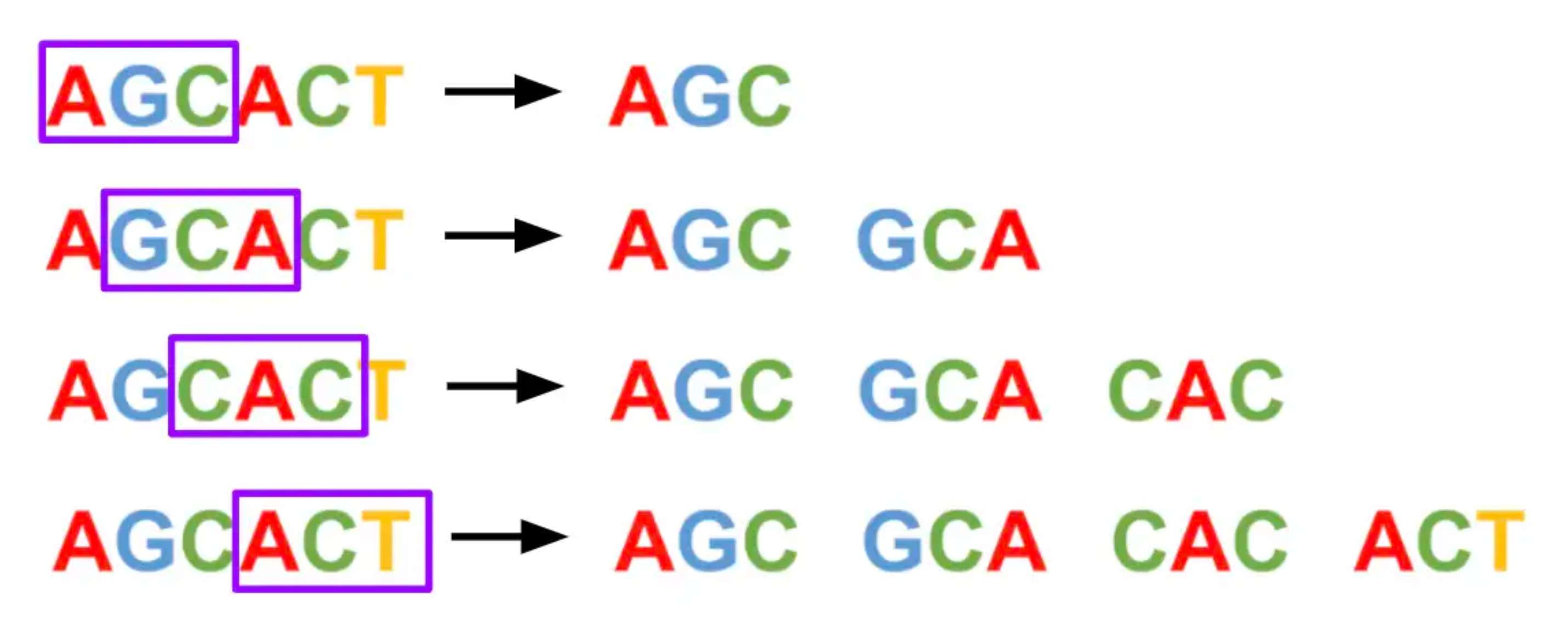
为什么不直接将AGCT编码为1 2 3 4进行序列的tokenization呢?而且这样占用的内存要小很多。
原因在于:功能序列一般都是多核苷酸或多氨基酸,k-mer技术可以将一段序列表示为1个数字,模型记住1个数字要比记住k个数字容易的多。
k-mer技术有两个可选值:窗口大小k值和k-mer的滑动步长
2. 手动实现一个简单的 Tokenizer
下面定义的函数 seq2kmer() 可以将 file_dir 文件中的序列转换为 kmers,并建立索引,粗略实现对序列的 tokenization。
def seq2kmer(file_dir, k):
"""
将file_dir文件中的序列转换为 kmers,滑动步长默认为1。
:param file_dir: 原始序列文件路径
:param k: 自定义窗口大小
:return: kmer文件和index文件
"""
"""读取示例数据,由于示例数据是多行的,为了避免换行符对结果产生影响,需要将多行字符串转换为单行字符串。"""
with open(file_dir) as object:
contents = object.read()
seq = contents.replace('\n', '') # contents.replace('\n', '') 将换行符全都去掉,将行与行紧密相连。
kmer = [seq[x:x + k] for x in range(len(seq) - k + 1)]
kmers = " ".join(kmer) # .join(kmer) 将列表kmer连接成一个字符串,kmer之间用空格分隔。
"""输出1:将转换后的kmers保存到txt文本中。"""
with open(f"{k}mers_{file_dir}", "w") as object:
object.write(kmers)
from itertools import product
nucleotides = 'ACGT'
"""生成所有kmers"""
kmers_all = [''.join(combination) for combination in product(nucleotides, repeat=k)] # 使用itertools.product生成所有可能的组合
"""输出2:穷举生成所有kmers,并保存到txt文本中。"""
with open(f"{k}mers_all.txt", "w") as object:
contents = str(kmers_all)
object.write(contents)
"""输出3:将file_dir文件的kmers索引写入txt文本"""
index = [kmers_all.index(seq[x:x + k]) for x in range(len(seq) - k + 1)]
with open(f"index_{k}mers_{file_dir}", "w") as object:
object.write(str(index))
# 使用示例数据运行seq2kmer
k = 6
seq2kmer("acrA.txt", k)
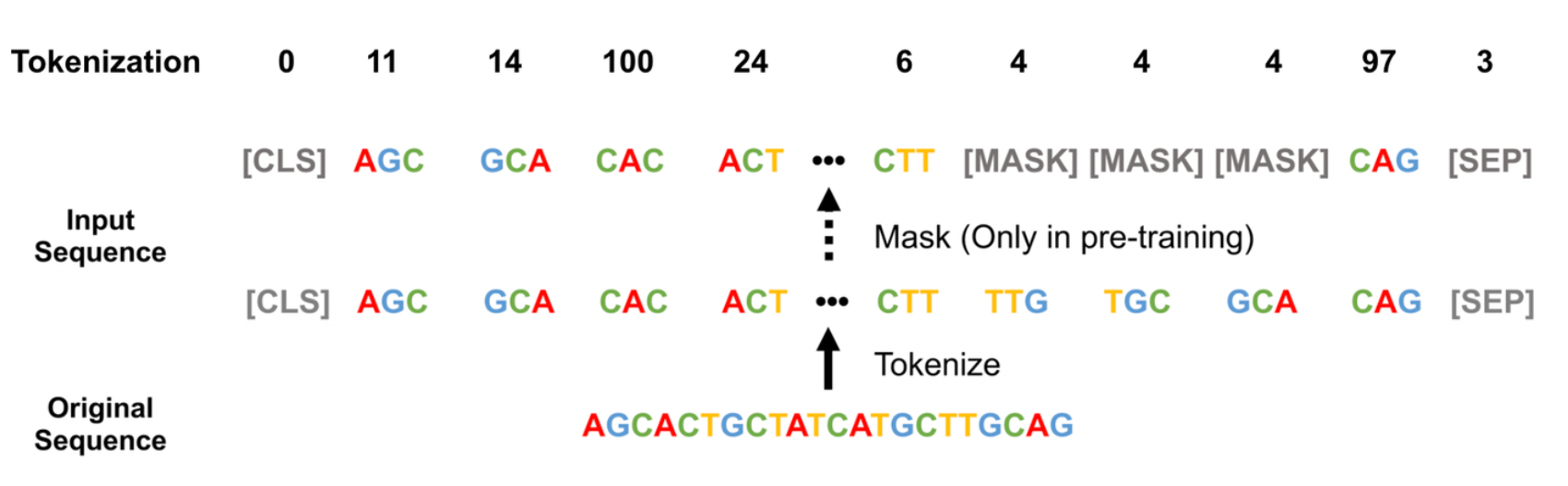
注意:以上代码暂未将 [CLS] 分类 token、[PAD] 填充 token、[UNK] 未知 token、[SEP] 分句 token 和 [MASK] 掩码 token (“[]” 也是 token 的一部分) 加入到词汇表中。
转换后的 k-mer 文件如下图所示:

为这些 kmers 建立的索引结果如下:

对于 6-mer,共计有 4096 ( 4 的 6 次方) 种可能的组合方式,再加上前面提到的 5 种特殊标记,所以 6-mer 的词汇表有 4111 种组合,索引范围为 [0, 4110] 。
3. 将 fasta 文件转变为预训练模型要求的数据输入格式:k-mer 格式
预训练模型要求的输入数据格式一般为kmer + label,如下图所示:

而我们手头的生物序列一般为fasta格式,下面定义的函数 fasta_tokenization() 可以将一个 fasta 格式多序列文本去掉标题行,然后一个序列归为一行,并将序列 k-mer 化。fasta_tokenization() 可以有效地将 fasta 格式生物序列处理为预训练模型要求的 k-mer 格式
def fasta_tokenization(fasta_file_dir, k):
"""
将fasta文件转换为k-mer
:param fasta_file_dir: 要处理的fasta文件路径
:param k: 自定义窗口大小
:return: f"cut_{fasta_file_dir} 和 f"{k}mers_{fasta_file_dir}"
"""
"""将fasta文件去掉标题行,并将每个序列合并为一行,然后保存到txt文本中"""
sequences = ""
with open("test.txt", "r") as object:
sequence = ""
for line in object:
line = line.strip()
if line.startswith(">"):
if sequence:
sequences = sequences + sequence + "\n"
sequence = ""
else:
sequence = sequence + line
if sequence:
sequences = sequences + sequence
"""
with open(file) as object: with open打开一个文件时,会返回一个文件对象object。使用for循环读取这个对象时,一次读取一行。
直接在字符串或者列表中使用for循环读取时,一次只能读取一个字符或列表单元。
所以,这里必须先将cut之后的序列保存为txt文本,然后再用with open打开,实现按行操作。
"""
with open(f"cut_{fasta_file_dir}", "w") as object:
object.write(sequences)
"""将fasta文件转换为kmers,并保存到txt文本中"""
with open(f"cut_{fasta_file_dir}") as object:
kmers_all = ""
for line in object:
kmer_line = [line[x:x + k] for x in range(len(line) - k + 1)]
kmers_line = " ".join(kmer_line)
kmers_all = kmers_all + kmers_line
with open(f"{k}mers_{fasta_file_dir}", "w") as object: # 也可直接保存为.tsv文件
object.write(kmers_all)
# 使用样例数据运行 fasta_tokenization()
fasta_file_dir = "test.txt"
fasta_tokenization("test.txt", 6)
本文由mdnice多平台发布















 2007
2007

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









