






分子动力学仿真软件Lammps最简单安装教程-Ubuntu20.04
- 1. 安装前的准备
- 2. 安装相关的依赖库
- 3. 编译Lammps
- 4. 根据需要安装额外的模块
1. 安装前的准备
1.1 安装好操作系统Ubuntu20.04, 其他版本也可以
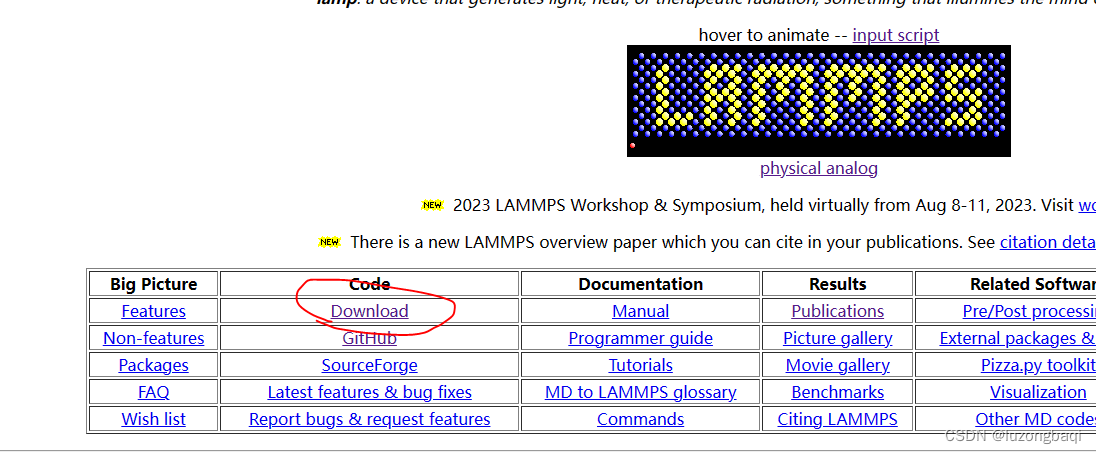
1.2 下载好Lammps源代码 下载地址
主界面中找到download --> 下载最新的稳定版本即可(本人下载)

1.3 下载并安装四个编译工具
sudo apt-get install gcc #安装gcc编译器
sudo apt-get install g++ #安装g++编译器
sudo apt-get install gfortran #安装gfortran编译器
sudo apt-get install make #安装make编译器
2. 安装相关的依赖库
Lammps源代码包解压后进入./src/MAKE/MACHINES,打开Makefile.ubuntu 查看所需要安装的库
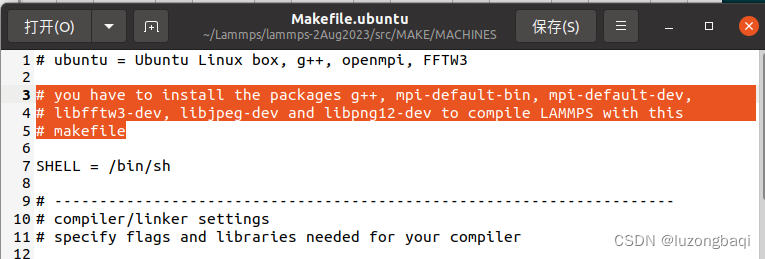
#安装相关的库
sudo apt-get install mpi-default-bin mpi-default-dev #MPICH
sudo apt-get install libfftw3-dev #FFTW
sudo apt-get install libjpeg-dev libpng-dev #有些人选择不安装,那就自己修改下make文件
3. 编译Lammps
进入src目录下开始编译
- 进入src目录,右键在终端打开
- 输入命令后编译
make mpi
- 可以在src目录下找到编译好的lmp_mpi (这个就是需要的东西)
- 将lmp_mpi 拷贝到指定文件夹下,并将改文件夹加入系统环境变量
#打开文本
sudo gedit ~/.bashrc
#在文本最后添加lmp_mpi所在的路径 例如:
export PATH=/home/admin1/Lammps/lammps-2Aug2023/bin:$PATH
- 运行测试
#书写一个案例后进入文件夹打开终端
# 单核运行,不使用MPI
lmp_serial -in in.file
# 并行计算,使用MPICH框架
mpirun -np 4 lmp_mpi -in in.file
4. 根据需要安装额外的模块
- 进入src目录下右键在终端打开
- 查看已经安装的额外模块
make package-status
- 根据需求安装/卸载模块
make yes-name #安装模块
make no-name #卸载模块
- 重新编译lammps
make mpi















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









