






KG4MM:融合知识图谱与多模态数据预测药物相互作用
知识图谱(Knowledge Graph, KG)作为表示不同实体间复杂关系的有效工具,已得到广泛应用。通过将信息编码为节点(实体)和边(关系),知识图谱简化了关联信息的追踪与分析。用于多模态学习的知识图谱(Knowledge Graphs for Multimodal Learning, KG4MM)借鉴了这一思想,利用知识图谱指导从图像和文本等多模态数据中进行学习。在 KG4MM 框架中,知识图谱充当结构化先验知识,引导模型在训练过程中关注每种数据模态中的关键信息。这种引导机制有助于模型聚焦于图像中最具区分性的特征以及文本中最具信息量的词汇。
在药物相互作用(Drug-Drug Interaction, DDI)预测领域,KG4MM 展现出显著优势。知识图谱结构能够将药物的分子图像表示和文本描述整合至统一框架内。这种整合视图通过同时捕获药物的化学结构信息和药理学背景知识,支持更精确的 DDI 预测。知识图谱为模型的预测过程提供了透明的推理路径,使得理解模型预测结果的依据更为便捷,增强了模型的可解释性。
本文旨在阐述 KG4MM 在 DDI 预测任务中的具体实现。文章将分步介绍知识图谱的构建过程,以及如何整合药物的分子结构信息和文本描述信息。通过具体实例,本文将演示知识图谱引导下的多模态学习如何应对医学与医疗保健研究中的实际挑战。其核心目标是展示 KG4MM 如何在真实的 DDI 预测任务中提升预测的准确性与可解释性。
方法论
KG4MM 方法论的核心在于将知识图谱置于整个处理流程的中心地位。知识图谱指导了每种数据模态的处理与理解方式。以药物相互作用预测为例,图谱中的每个药物节点均关联两种信息模态:一种是由其 SMILES (Simplified Molecular Input Line Entry System) 化学式衍生的分子图像,另一种是包含其类别、官能团及其他关键属性的文本描述。
KG4MM 的独特之处在于利用图神经网络(Graph Neural Network, GNN)将知识图谱的结构信息与多模态数据进行有效连接。GNN 通过分析药物在图谱中的拓扑位置(即其与其他实体如蛋白质、疾病等的连接关系),来判断其对应图像的哪些区域和文本描述中的哪些词语应获得更高的关注度。图谱中的边(例如,表示药物与蛋白质的结合关系、与疾病的治疗关系等)为 GNN 提供了依据,使其能够判断哪些视觉和文本特征具有更高的重要性。因此,知识图谱不仅提供了上下文信息,更主动地引导模型的注意力聚焦于最具信息量的数据元素。
KG4MM 的优势在于结合了神经网络的模式识别能力与知识图谱的显式关系表示能力。GNN 特别适用于从连接数据中学习,使得模型能够基于已有的药物相互作用知识和生化特性知识进行构建。这种知识引导的学习范式不仅提升了预测准确率,而且通过明确指示影响预测结果的具体图谱连接,产生了清晰、可解释的预测结果。
实现概述
该系统的核心是一个集成了所有组件的中央知识图谱。该图谱捕获了药物、蛋白质和疾病之间的有向关系,例如药物“结合到(binds_to)”蛋白质、“抑制(inhibits)”靶点或“治疗(treats)”疾病等。将知识图谱置于设计的核心,使得处理流程的每一步都能利用其结构化的医学知识。
在数据准备阶段,每个药物节点关联两种表示形式。第一种是利用 RDKit 库从药物的 SMILES 化学式生成的分子图像。第二种是文本描述,概括了药物的类别、官能团及其他相关细节。图像和文本均直接链接到图谱中对应的药物节点,确保视觉和语言特征与底层的知识结构保持一致。
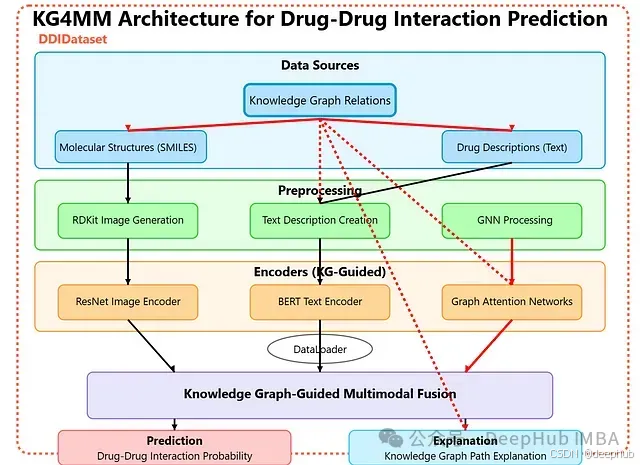
知识图谱本身的表示学习依赖于图卷积网络(Graph Convolutional Network, GCN)。GCN 从每个节点的拓扑位置及其在图谱中的连接关系中学习,生成能够编码药物、蛋白质和疾病之间相互关系的嵌入向量(embedding)。与此同时,多模态编码器负责将图像和文本转换为特征向量:采用 ResNet 模型处理分子图像,而采用 BERT 模型转换文本描述。
最后,图注意力网络(Graph Attention Network, GAT)负责融合知识图谱嵌入与视觉、文本特征。注意力机制利用图谱结构来加权来自不同模态的最重要特征。融合后的表示被输入到一个预测模块,该模块最终判断两种药物之间是否存在相互作用。同时,注意力权重揭示了哪些图谱连接、图像区域或文本片段对模型的决策贡献最大,从而为每个预测提供了清晰的解释。
详细实现
实现过程首先需要安装并导入必要的 Python 库。此步骤确保所有用于深度学习(PyTorch, torchvision)、图谱处理(NetworkX, torch-geometric)、化学信息学(RDKit, OpenBabel)以及文本编码(HuggingFace Transformers)的软件包在环境中可用。此外,还包括 pandas、NumPy 和 Matplotlib 等基础支持库。安装完成后,导入所需的库和模块,以便在后续代码单元中使用。
# 安装必要的包
!pip install torch torchvision transformers networkx spacy rdflib rdkit pillow scikit-learn matplotlib seaborn torch-geometric
# pip 安装失败
!apt-get install openbabel
!pip install openbabel-wheel
# 导入库
import torch
import torch.nn as nn
from torch.utils.data import Dataset, DataLoader
import torchvision.models as models
import torchvision.transforms as transforms
from transformers import BertModel, BertTokenizer
import networkx as nx
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import json
import os
from rdkit import Chem
from rdkit.Chem import Draw
from PIL import Image
import io
import base64
from openbabel import openbabel
from torch_geometric.data import Data
import torch_geometric.nn as geom_nn
接下来,创建一个目录用于存储生成的药物分子图像。然后从公共存储库下载简化的 DrugBank 样本数据集,并将其保存为 TSV (Tab-Separated Values) 文件。该文件随后被加载到 pandas DataFrame 中,形成一个包含每种药物的唯一标识符、名称、分子结构的 InChI (International Chemical Identifier) 字符串以及描述性元数据(如类别和组别)的表格。这个结构化的数据集为后续生成视觉和文本表示提供了基础。
# 创建用于数据存储的目录
!mkdir -p data/drug_images
# 下载 DrugBank 样本数据(用于演示的简化版本)
!wget -q -O data/drugbank_sample.tsv https://raw.githubusercontent.com/dhimmel/drugbank/gh-pages/data/drugbank-slim.tsv
# 加载 DrugBank 数据
drug_df = pd.read_csv('data/drugbank_sample.tsv', sep='\t')
原始数据集中提供的分子结构信息为 InChI 格式。为了便于后续处理,特别是使用 RDKit 生成图像,需要将 InChI 格式转换为 SMILES 格式。SMILES 提供了一种简洁的、基于文本的化学结构表示方法。以下代码段展示了如何使用 OpenBabel 库执行此转换。
# 通过将 InChI 转换为 SMILES 来创建 SMILES 列
def inchi_to_smiles_openbabel(inchi_str):
try:
# 从 InChI 创建 Open Babel OBMol 对象
obConversion = openbabel.OBConversion()
obConversion.SetInAndOutFormats("inchi", "smiles")
mol = openbabel.OBMol()
# 将 InChI 转换为分子
# 同时移除多余的换行符或空格
if obConversion.ReadString(mol, inchi_str):
return obConversion.WriteString(mol).strip()
else:
return None
except Exception as e:
print(f"Error converting InChI to SMILES: {inchi_str}. Error: {e}")
return None
# 将转换应用于数据帧中的每个 InChI
drug_df['smiles'] = drug_df['inchi'].apply(inchi_to_smiles_openbabel)
知识图谱构建。 系统构建了一个有向的医学知识图谱,旨在捕获药物、蛋白质和疾病之间的相互关系。图谱中的每个节点代表一个实体(药物、蛋白质或疾病),每条边则编码一种特定的相互作用或关系,例如 binds_to(结合到)、inhibits(抑制)或 treats(治疗)。这些连接蕴含了关于药物如何影响生物靶点和疾病状态的领域专家知识。
该知识图谱作为结构化关系信息的来源,模型将其与图像和文本特征相结合进行学习。通过显式地表示领域知识,知识图谱不仅增强了 DDI 预测的准确性,也提升了解释两种药物可能发生相互作用原因的能力。
# 初始化医学知识图谱
medical_kg = nx.DiGraph()
# 从 DrugBank 提取药物实体
# 限制为 50 种药物用于演示
drug_entities = drug_df['name'].dropna().unique().tolist()[:50]
# 创建药物节点
for drug in drug_entities:
medical_kg.add_node(drug, type='drug')
# 添加生物医学实体(蛋白质、靶点、疾病)
protein_entities = ["Cytochrome P450", "Albumin", "P-glycoprotein", "GABA Receptor",
"Serotonin Receptor", "Beta-Adrenergic Receptor", "ACE", "HMGCR"]
disease_entities = ["Hypertension", "Diabetes", "Depression", "Epilepsy",
"Asthma", "Rheumatoid Arthritis", "Parkinson's Disease"]
for protein in protein_entities:
medical_kg.add_node(protein, type='protein')
for disease in disease_entities:
medical_kg.add_node(disease, type='disease')
# 添加关系(基于常见的药物机制和相互作用)
# 药物-蛋白质关系
drug_protein_relations = [
("Warfarin", "binds_to", "Albumin"),
("Atorvastatin", "inhibits", "HMGCR"),
("Diazepam", "modulates", "GABA Receptor"),
("Fluoxetine", "inhibits", "Serotonin Receptor"),
("Phenytoin", "induces", "Cytochrome P450"),
("Metoprolol", "blocks", "Beta-Adrenergic Receptor"),
("Lisinopril", "inhibits", "ACE"),
("Rifampin", "induces", "P-glycoprotein"),
("Carbamazepine", "induces", "Cytochrome P450"),
("Verapamil", "inhibits", "P-glycoprotein")
]
# 药物-疾病关系
drug_disease_relations = [
("Lisinopril", "treats", "Hypertension"),
("Metformin", "treats", "Diabetes"),
("Fluoxetine", "treats", "Depression"),
("Phenytoin", "treats", "Epilepsy"),
("Albuterol", "treats", "Asthma"),
("Methotrexate", "treats", "Rheumatoid Arthritis"),
("Levodopa", "treats", "Parkinson's Disease")
]
# 已知的药物-药物相互作用(基于实际医学知识)
drug_drug_interactions = [
("Goserelin", "interacts_with", "Desmopressin", "increases_anticoagulant_effect"),
("Goserelin", "interacts_with", "Cetrorelix", "increases_bleeding_risk"),
("Cyclosporine", "interacts_with", "Felypressin", "decreases_efficacy"),
("Octreotide", "interacts_with", "Cyanocobalamin", "increases_hypoglycemia_risk"),
("Tetrahydrofolic acid", "interacts_with", "L-Histidine", "increases_statin_concentration"),
("S-Adenosylmethionine", "interacts_with", "Pyruvic acid", "decreases_efficacy"),
("L-Phenylalanine", "interacts_with", "Biotin", "increases_sedation"),
("Choline", "interacts_with", "L-Lysine", "decreases_efficacy")
]
# 将所有关系添加到知识图谱
for s, r, o in drug_protein_relations:
if s in medical_kg and o in medical_kg:
medical_kg.add_edge(s, o, relation=r)
for s, r, o in drug_disease_relations:
if s in medical_kg and o in medical_kg:
medical_kg.add_edge(s, o, relation=r)
for s, r, o, mechanism in drug_drug_interactions:
if s in medical_kg and o in medical_kg:
medical_kg.add_edge(s, o, relation=r, mechanism=mechanism)
多模态数据处理。 系统中每种药物由三种互补的数据类型表示。首先, SMILES 表示被转换为 RDKit 分子对象,并渲染成二维分子结构图像。
# 使用 RDKit 生成分子结构图像的函数
def generate_molecule_image(smiles_string, size=(224, 224)):
try:
mol = Chem.MolFromSmiles(smiles_string)
if mol:
img = Draw.MolToImage(mol, size=size)
return img
else:
return None
except:
return None
然后通过整合药物的名称、类别、组别信息以及其他可用的元数据,构建结构化的文本描述。
# 创建结合各种信息的药物文本描述的函数
def create_drug_description(row):
description = f"Drug name: {row['name']}. "
if pd.notna(row.get('category')):
description += f"Category: {row['category']}. "
if pd.notna(row.get('groups')):
description += f"Groups: {row['groups']}. "
if pd.notna(row.get('description')):
description += f"Description: {row['description']}"
# 返回完整的描述
return description.strip() # 添加 strip() 移除末尾可能存在的空格
最后进行知识图谱嵌入。此过程旨在将图谱中的节点(实体)和边(关系)映射到低维向量空间。常用的方法如 TransE 或基于 GNN 的嵌入方法,通过优化目标函数使得向量表示能够捕捉图谱的结构信息。例如对于一个三元组(头实体 h, 关系 r, 尾实体 t),目标是使 h + r ≈ t。经过训练,生成的嵌入向量能够反映实体间的语义关系和图谱的整体拓扑结构。这些嵌入向量将作为图谱结构信息的数值表示,输入到后续模型中。为了适配现代 GNN 框架(如 PyTorch Geometric, PyG),需要将 NetworkX 图对象转换为 PyG 的 Data 对象格式。
# 将 NetworkX 图转换为 PyG 图,用于现代图神经网络处理
def convert_nx_to_pyg(nx_graph):
# 创建节点映射
node_to_idx = {node: i for i, node in enumerate(nx_graph.nodes())}
# 创建边列表
src_nodes = []
dst_nodes = []
edge_types = []
edge_type_to_idx = {}
for u, v, data in nx_graph.edges(data=True):
relation = data.get('relation', 'unknown')
if relation not in edge_type_to_idx:
edge_type_to_idx[relation] = len(edge_type_to_idx)
src_nodes.append(node_to_idx[u])
dst_nodes.append(node_to_idx[v])
edge_types.append(edge_type_to_idx[relation])
# 创建 PyG 图
edge_index = torch.tensor([src_nodes, dst_nodes], dtype=torch.long)
edge_type = torch.tensor(edge_types, dtype=torch.long)
# 创建节点特征
node_types = []
for node in nx_graph.nodes():
node_type = nx_graph.nodes[node].get('type', 'unknown')
node_types.append(node_type)
# 对节点类型进行独热编码
unique_node_types = sorted(set(node_types))
node_type_to_idx = {nt: i for i, nt in enumerate(unique_node_types)}
node_type_features = torch.zeros(len(node_types), len(unique_node_types))
for i, nt in enumerate(node_types):
node_type_features[i, node_type_to_idx[nt]] = 1.0
# 使用正确的属性创建 PyG Data 对象
g = Data(
edge_index=edge_index,
edge_type=edge_type,
x=node_type_features # PyG 中的节点特征存储在 'x' 中
)
# 创建反向映射以供后续使用
idx_to_node = {idx: node for node, idx in node_to_idx.items()}
idx_to_edge_type = {idx: edge_type for edge_type, idx in edge_type_to_idx.items()}
return g, node_to_idx, idx_to_node, edge_type_to_idx, idx_to_edge_type
# 将 medical_kg 转换为 PyG 图
pyg_graph, node_to_idx, idx_to_node, edge_type_to_idx, idx_to_edge_type = convert_nx_to_pyg(medical_kg)
这些视觉、文本和结构化表示被整合处理,以便模型能够融合它们进行药物相互作用的预测。
# 处理药物数据以创建多模态表示
drug_data = []
for idx, row in drug_df.iterrows():
# 确保药物在 KG 中且有 SMILES 数据
if row['name'] in node_to_idx and pd.notna(row.get('smiles')):
# 生成分子图像
img = generate_molecule_image(row['smiles'])
if img:
# 确保目录存在
img_dir = "data/drug_images"
os.makedirs(img_dir, exist_ok=True)
img_path = os.path.join(img_dir, f"{row['drugbank_id']}.png")
img.save(img_path)
# 创建文本描述
description = create_drug_description(row)
# 存储药物信息
drug_data.append({
'id': row['drugbank_id'],
'name': row['name'],
'smiles': row['smiles'],
'description': description,
'image_path': img_path
})
drug_data_df = pd.DataFrame(drug_data)
编码器开发。 MultimodalNodeEncoder 模块负责将每个节点的分子图像和文本描述转换为维度兼容的特征向量。它首先利用预训练的深度卷积网络(如 ResNet)处理分子图像,提取其视觉特征表示。同时,采用预训练的语言模型(如 BERT)处理药物的文本描述,提取其语义特征表示。随后,通过线性投影层将两种模态的特征向量映射到相同的低维空间,确保视觉和文本信号可以在知识图谱结构的指导下进行有意义的融合。
# 处理节点的视觉和文本特征
class MultimodalNodeEncoder(nn.Module):
def __init__(self, output_dim=128):
super(MultimodalNodeEncoder, self).__init__()
# 图像编码器 (ResNet)
resnet = models.resnet18(pretrained=True)
# 移除最后的完全连接层以获得 512 个特征
self.image_encoder = nn.Sequential(*list(resnet.children())[:-1])
self.image_projection = nn.Linear(512, output_dim)
# 文本编码器 (BERT)
self.tokenizer = BertTokenizer.from_pretrained('bert-base-uncased')
self.text_encoder = BertModel.from_pretrained('bert-base-uncased')
# BERT base 输出 768 个特征
self.text_projection = nn.Linear(768, output_dim)
def forward(self, image, text):
# 图像编码
# 确保输入图像是 4D 张量 (B, C, H, W)
if image.dim() == 3:
image = image.unsqueeze(0) # 添加批次维度
img_features = self.image_encoder(image).squeeze(-1).squeeze(-1) # (B, 512)
img_features = self.image_projection(img_features) # (B, output_dim)
# 文本编码
# 确保 text 是一个列表或可迭代对象
if isinstance(text, str):
text = [text]
encoded_input = self.tokenizer(text, padding=True, truncation=True,
return_tensors="pt", max_length=128)
# 将编码后的输入移动到与图像相同的设备
input_ids = encoded_input['input_ids'].to(image.device)
attention_mask = encoded_input['attention_mask'].to(image.device)
text_outputs = self.text_encoder(input_ids=input_ids,
attention_mask=attention_mask)
# 使用 [CLS] 标记嵌入(第一个标记)
text_features = text_outputs.last_hidden_state[:, 0, :] # (B, 768)
text_features = self.text_projection(text_features) # (B, output_dim)
return img_features, text_features
模型集成。 KGGuidedMultimodalModel 在知识图谱的引导下,融合每个节点的视觉、文本和类型嵌入,以预测药物-药物相互作用。该模型首先将每个节点的图像和文本特征投影到一个共享的嵌入空间,并为其节点类型(如药物、蛋白质、疾病)分配一个独立的嵌入向量。然后,利用 GNN 层(如 GCN)在知识图谱上传播这些嵌入,使得每个节点能够聚合自身特征及其邻居节点的信号。接着,采用图注意力网络(GAT)层,根据连接强度和类型对聚合后的特征进行加权。在评估药物对时,模型获取它们经过 GNN 和 GAT 精炼后的节点表示,通过拼接、逐元素乘积和差值等方式组合这两个表示,并将结果输入到一个预测头(通常是多层感知机),最终输出相互作用的概率。通过让知识图谱的拓扑结构指导多模态信号的融合过程,该模型生成的预测结果不仅准确,而且其推理过程可追溯至底层的图谱结构。
# 定义 KG 引导的多模态模型
class KGGuidedMultimodalModel(nn.Module):
def __init__(self, pyg_graph, num_node_types, num_edge_types, node_to_idx, idx_to_node, hidden_dim=128):
super(KGGuidedMultimodalModel, self).__init__()
self.pyg_graph = pyg_graph # PyG Data object
self.node_to_idx = node_to_idx
self.idx_to_node = idx_to_node
self.hidden_dim = hidden_dim
# 用于处理节点关联数据的多模态编码器
self.multimodal_encoder = MultimodalNodeEncoder(output_dim=hidden_dim)
# 节点类型嵌入 (使用图谱中提供的 one-hot 编码特征维度)
self.node_type_projection = nn.Linear(num_node_types, hidden_dim)
# 用于知识图谱处理的图神经网络层(PyG GCNConv)
self.gnn_layers = nn.ModuleList([
geom_nn.GCNConv(hidden_dim, hidden_dim),
geom_nn.GCNConv(hidden_dim, hidden_dim),
])
# 用于将多模态特征与图谱结构集成的图注意力网络(PyG GATConv)
num_heads = 4
# GAT 输出维度是 hidden_dim * num_heads, 需要调整后续层或 GAT 输出
self.gat_layer = geom_nn.GATConv(hidden_dim, hidden_dim // num_heads, heads=num_heads)
# 关系预测层 - 输入维度需要匹配 GAT 输出和特征组合方式
# 组合特征维度: hidden_dim (drug1) + hidden_dim (drug2) + hidden_dim (element-wise product) + hidden_dim (abs diff) = hidden_dim * 4
self.relation_prediction = nn.Sequential(
nn.Linear(hidden_dim * 4, hidden_dim * 2), # GAT 输出是 hidden_dim
nn.ReLU(),
nn.Dropout(0.3),
nn.Linear(hidden_dim * 2, hidden_dim),
nn.ReLU(),
nn.Dropout(0.3),
nn.Linear(hidden_dim, 1)
)
def get_initial_node_embeddings(self, drug_data_df):
# 初始化所有节点的嵌入
num_nodes = self.pyg_graph.x.size(0)
device = self.pyg_graph.x.device
node_embeddings = torch.zeros((num_nodes, self.hidden_dim), device=device)
# 处理节点类型特征
node_type_features = self.pyg_graph.x # (num_nodes, num_node_types)
node_embeddings += self.node_type_projection(node_type_features)
# 处理有图像和文本的药物节点
drug_indices = []
images = []
texts = []
drug_name_to_node_idx = {}
for i, row in drug_data_df.iterrows():
if row['name'] in self.node_to_idx:
node_idx = self.node_to_idx[row['name']]
drug_name_to_node_idx[row['name']] = node_idx
drug_indices.append(node_idx)
# 加载图像 (需要预处理)
try:
img = Image.open(row['image_path']).convert('RGB')
# 应用与训练时相同的变换
transform = transforms.Compose([
transforms.Resize((224, 224)),
transforms.ToTensor(),
transforms.Normalize(mean=[0.485, 0.456, 0.406], std=[0.229, 0.224, 0.225])
])
images.append(transform(img))
except Exception as e:
print(f"Warning: Could not load image for {row['name']}: {e}")
images.append(torch.zeros((3, 224, 224))) # Placeholder
texts.append(row['description'])
if images:
images_tensor = torch.stack(images).to(device)
# 批量处理图像和文本
img_feats, text_feats = self.multimodal_encoder(images_tensor, texts) # (num_drugs, hidden_dim)
# 将多模态特征添加到对应节点的嵌入中 (可以采用加法或更复杂的融合)
# 这里使用加法作为示例
node_embeddings[drug_indices] += (img_feats + text_feats) / 2 # 平均融合
return node_embeddings, drug_name_to_node_idx
def forward(self, drug1_name, drug2_name, initial_node_embeddings, drug_name_to_node_idx):
# 使用预先计算的初始节点嵌入
x = initial_node_embeddings
edge_index = self.pyg_graph.edge_index
# 应用图卷积来传播信息
for layer in self.gnn_layers:
x = layer(x, edge_index)
x = torch.relu(x) # (num_nodes, hidden_dim)
# 应用图注意力来集成特征
# GATConv 输出维度是 (num_nodes, heads * out_channels) = (num_nodes, hidden_dim)
x = self.gat_layer(x, edge_index)
# GATConv 通常后接激活函数和可能的 Dropout
x = torch.relu(x)
# 获取两种药物的最终表示
drug1_node_idx = drug_name_to_node_idx.get(drug1_name)
drug2_node_idx = drug_name_to_node_idx.get(drug2_name)
# 处理药物不在映射中的情况 (例如,如果 drug_data_df 不完整)
if drug1_node_idx is None or drug2_node_idx is None:
print(f"Warning: Drug node index not found for {drug1_name} or {drug2_name}. Returning zero probability.")
# 返回一个表示低概率的张量,确保维度匹配
return torch.tensor(0.0, device=x.device)
drug1_repr = x[drug1_node_idx] # (hidden_dim)
drug2_repr = x[drug2_node_idx] # (hidden_dim)
# 预测相互作用
# 以多种方式组合表示以捕获关系
concat_repr = torch.cat([
drug1_repr,
drug2_repr,
drug1_repr * drug2_repr, # 逐元素乘积
torch.abs(drug1_repr - drug2_repr) # 绝对差值
], dim=0) # (hidden_dim * 4)
# 确保输入维度与 prediction layer 匹配
interaction_prob = torch.sigmoid(self.relation_prediction(concat_repr)) # 输出单个概率值
return interaction_prob.squeeze() # 移除多余维度
知识提取。 为了解释模型对特定药物对相互作用的预测,需要从完整的知识图谱中提取与该药物对相关的子图。该过程首先检查这对药物之间是否存在直接的边(表示已知的直接相互作用),并记录其属性(如相互作用机制)。其次,识别与这两种药物均有连接的共享节点(如共同作用的蛋白质靶点或共同治疗的疾病),这可能揭示间接相互作用的机制。最后,查找连接这两种药物的所有达到预定长度阈值的简单路径,以发现通过中间实体(其他药物、蛋白质等)的间接关联。提取出的子图由关键节点和边构成,它捕获了支持预测相互作用的领域知识,并有助于下游解释模块识别和强调最相关的多模态特征。
# 检索与药物对相关的知识子图的函数
def retrieve_knowledge_subgraph(graph, drug1, drug2, max_path_length=3):
relevant_knowledge = {
'direct_interaction': None,
'common_targets': [],
'paths': []
}
# 检查节点是否存在于图中
if drug1 not in graph or drug2 not in graph:
print(f"Warning: One or both drugs ({drug1}, {drug2}) not found in the knowledge graph.")
return relevant_knowledge
# 检查直接相互作用 (双向)
if graph.has_edge(drug1, drug2):
relevant_knowledge['direct_interaction'] = graph.get_edge_data(drug1, drug2)
elif graph.has_edge(drug2, drug1):
relevant_knowledge['direct_interaction'] = graph.get_edge_data(drug2, drug1) # 考虑反向边
# 查找共同邻居(潜在的共同靶点或相关实体)
try:
drug1_neighbors = set(graph.neighbors(drug1))
drug2_neighbors = set(graph.neighbors(drug2))
common_neighbors = drug1_neighbors.intersection(drug2_neighbors)
for common_node in common_neighbors:
node_type = graph.nodes[common_node].get('type', 'unknown')
# 可以根据需要筛选特定类型的共同节点
# if node_type == 'protein' or node_type == 'disease':
relevant_knowledge['common_targets'].append(common_node)
except nx.NetworkXError as e:
print(f"Error finding common neighbors for {drug1} and {drug2}: {e}")
# 查找药物之间的路径(最多 max_path_length)
try:
# 查找双向路径
paths1 = list(nx.all_simple_paths(graph, source=drug1, target=drug2, cutoff=max_path_length))
paths2 = list(nx.all_simple_paths(graph, source=drug2, target=drug1, cutoff=max_path_length))
# 合并路径并去重(注意路径方向)
relevant_knowledge['paths'] = paths1 + [p[::-1] for p in paths2] # 将反向路径也视为连接路径
except nx.NodeNotFound:
# 节点不在图中(理论上前面已检查,但作为保险)
pass
except nx.NetworkXNoPath:
# 路径不存在
pass
return relevant_knowledge
自定义批次整理(Collate Function)。 在使用 DataLoader 时,需要一个自定义的 collate_fn 来处理数据集中可能存在的无效样本(例如,由于图像加载失败)。此函数首先过滤掉批次中值为 None 的样本。然后,它将剩余有效样本中的药物对图像堆叠成批处理张量,并将相应的文本描述、药物名称收集到列表中。相互作用标签同样被组合成单个张量。该函数返回一个包含批处理后各组件的字典。如果整个批次都无效,则返回包含空张量和空列表的占位符字典,确保模型输入的一致性和健壮性,即使在存在异构或缺失数据的情况下也能稳定运行。
# 自定义整理函数以处理 None 值
def custom_collate_fn(batch):
# 过滤掉 None 值 (由 Dataset 的 __getitem__ 返回)
batch = [item for item in batch if item is not None]
# 如果过滤后批次为空,则返回特殊标记或空字典
if not batch:
return None # 或者返回一个表示空批次的结构
# 处理非 None 项
drug1_imgs = torch.stack([item['drug1_img'] for item in batch])
drug1_texts = [item['drug1_text'] for item in batch]
drug1_names = [item['drug1_name'] for item in batch]
drug2_imgs = torch.stack([item['drug2_img'] for item in batch])
drug2_texts = [item['drug2_text'] for item in batch]
drug2_names = [item['drug2_name'] for item in batch]
labels = torch.stack([item['label'] for item in batch])
return {
'drug1_img': drug1_imgs,
'drug1_text': drug1_texts,
'drug1_name': drug1_names,
'drug2_img': drug2_imgs,
'drug2_text': drug2_texts,
'drug2_name': drug2_names,
'label': labels
}
数据集准备。 为了训练 DDI 预测模型,需要构建一个包含正负样本的数据集。该过程首先从已知的药物相互作用数据(drug_drug_interactions)中提取正样本对。然后,通过随机采样药物对来生成等量的负样本(确保采样到的药物对不存在于已知相互作用中),以实现类别平衡。DDIDataset 类负责此任务。在其 __getitem__ 方法中,对于给定的索引,它检索对应的药物对名称和标签,加载并预处理这两种药物的分子图像(应用图像变换)和文本描述。如果任何药物的数据(特别是图像)加载失败,则该样本被标记为无效(返回 None),由 custom_collate_fn 处理。最终,每个有效样本以字典形式返回,包含药物对的图像张量、文本描述、名称以及二元相互作用标签。这种方式确保了数据集为模型训练提供了结构一致、经过预处理且包含正负样本的批次数据。
# 定义用于 DDI 预测的数据集
class DDIDataset(Dataset):
def __init__(self, drug_data_df, drug_drug_interactions, medical_kg, node_to_idx, transform=None):
# 确保 drug_data_df 包含必要的列: 'name', 'image_path', 'description'
self.drug_data = drug_data_df[drug_data_df['name'].isin(node_to_idx)].reset_index(drop=True)
self.drug_name_to_data_idx = {row['name']: i for i, row in self.drug_data.iterrows()}
self.node_to_idx = node_to_idx # KG node index mapping
self.transform = transform or transforms.Compose([
transforms.Resize((224, 224)),
transforms.ToTensor(),
transforms.Normalize(mean=[0.485, 0.456, 0.406], std=[0.229, 0.224, 0.225])
])
# 创建带有相互作用标签的药物对
self.pairs = []
# 只考虑在 drug_data (有图像/文本) 和 KG 中都存在的药物
available_drug_names = list(self.drug_name_to_data_idx.keys())
# 正样本(已知的相互作用)
positive_pairs_set = set()
for interaction in drug_drug_interactions:
drug1, _, drug2, _ = interaction
# 确保两个药物都在可用列表中
if drug1 in available_drug_names and drug2 in available_drug_names:
# 1 表示正相互作用
self.pairs.append((drug1, drug2, 1))
positive_pairs_set.add(tuple(sorted((drug1, drug2)))) # 使用排序后的元组以避免重复和方向问题
# 生成负样本
np.random.seed(42)
neg_count = 0
max_neg = len(self.pairs) # 目标负样本数量等于正样本数量
attempts = 0 # 防止无限循环
max_attempts = max_neg * 10
while neg_count < max_neg and attempts < max_attempts:
attempts += 1
# 从可用药物中随机选择两个不同的药物
if len(available_drug_names) < 2: break # 不足以选择两个药物
i, j = np.random.choice(len(available_drug_names), 2, replace=False)
drug1, drug2 = available_drug_names[i], available_drug_names[j]
# 检查是否为已知正样本 (无方向)
pair_tuple = tuple(sorted((drug1, drug2)))
if pair_tuple not in positive_pairs_set:
# 0 表示负相互作用
self.pairs.append((drug1, drug2, 0))
neg_count += 1
# 将负样本也加入集合,防止重复添加
positive_pairs_set.add(pair_tuple)
if neg_count < max_neg:
print(f"Warning: Could only generate {neg_count} negative samples (target: {max_neg}).")
def __len__(self):
return len(self.pairs)
def __getitem__(self, idx):
try:
drug1_name, drug2_name, label = self.pairs[idx]
# 获取 drug1 数据索引
drug1_data_idx = self.drug_name_to_data_idx[drug1_name]
drug1_data = self.drug_data.iloc[drug1_data_idx]
# 加载 drug1 图像并进行错误处理
try:
drug1_img = Image.open(drug1_data['image_path']).convert('RGB')
if self.transform:
drug1_img = self.transform(drug1_img)
except Exception as e:
# print(f"Error loading image for {drug1_name} at index {idx}: {str(e)}. Skipping sample.")
return None # 返回 None,由 collate_fn 处理
drug1_text = drug1_data['description']
# 获取 drug2 数据索引
drug2_data_idx = self.drug_name_to_data_idx[drug2_name]
drug2_data = self.drug_data.iloc[drug2_data_idx]
# 加载 drug2 图像并进行错误处理
try:
drug2_img = Image.open(drug2_data['image_path']).convert('RGB')
if self.transform:
drug2_img = self.transform(drug2_img)
except Exception as e:
# print(f"Error loading image for {drug2_name} at index {idx}: {str(e)}. Skipping sample.")
return None # 返回 None
drug2_text = drug2_data['description']
return {
'drug1_img': drug1_img, # Tensor
'drug1_text': drug1_text, # str
'drug1_name': drug1_name, # str
'drug2_img': drug2_img, # Tensor
'drug2_text': drug2_text, # str
'drug2_name': drug2_name, # str
'label': torch.tensor(label, dtype=torch.float32) # Tensor
}
except Exception as e:
# print(f"General error in __getitem__ for index {idx}: {str(e)}. Skipping sample.")
return None # 捕获其他潜在错误
模型训练。 模型训练过程在一个预设的轮数(epochs)内迭代进行。首先,将模型及其关联的知识图谱数据(PyG Data 对象)移动到指定的计算设备(GPU 或 CPU)。每个轮次包含训练阶段和验证阶段。在训练阶段,模型设置为训练模式(model.train())。数据加载器按批次提供药物对数据。对于每个非空批次,模型接收药物对的图像、文本和名称,计算初始节点嵌入,然后通过 GNN 和 GAT 层进行前向传播,最终输出相互作用概率。使用二元交叉熵(Binary Cross-Entropy, BCE)损失函数计算预测概率与真实标签之间的损失。然后,通过反向传播计算梯度,并使用 Adam 优化器更新模型参数。同时记录每个批次的损失和预测准确的数量。轮次结束时,计算并报告平均训练损失和训练准确率。在验证阶段,模型切换到评估模式(model.eval()),不计算梯度。使用验证数据加载器重复前向传播过程,计算并报告平均验证损失和验证准确率。这个过程允许监控模型的学习进度和泛化能力。
# 训练函数
def train_kg4mm_model(model, train_loader, val_loader, drug_data_df, epochs=5):
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
print(f"Using device: {device}")
model = model.to(device)
# 将图谱数据也移动到设备
model.pyg_graph = model.pyg_graph.to(device)
criterion = nn.BCELoss()
optimizer = torch.optim.Adam(model.parameters(), lr=0.0001) # 调整学习率
for epoch in range(epochs):
# --- 训练阶段 ---
model.train()
train_loss = 0
train_correct = 0
train_samples = 0
# 在每个 epoch 开始时计算一次初始嵌入
# 注意:如果节点特征会变化,则需要在每次迭代中重新计算或更新
initial_node_embeddings, drug_name_to_node_idx = model.get_initial_node_embeddings(drug_data_df)
initial_node_embeddings = initial_node_embeddings.to(device) # 确保在正确设备上
print(f"\nEpoch {epoch+1}/{epochs} - Training...")
for i, batch in enumerate(train_loader):
# 跳过空批次 (由 collate_fn 返回 None)
if batch is None:
print(f"Skipping empty batch {i+1}")
continue
# 从批次中提取数据 (已经在 collate_fn 中处理)
drug1_names = batch['drug1_name']
drug2_names = batch['drug2_name']
labels = batch['label'].to(device) # (batch_size)
optimizer.zero_grad()
# 批量处理预测
batch_outputs = []
valid_indices = [] # 跟踪成功处理的样本索引
for j in range(len(drug1_names)):
drug1_name = drug1_names[j]
drug2_name = drug2_names[j]
# 检查药物是否在映射中 (模型内部也会检查,但这里可以提前跳过)
if drug1_name not in drug_name_to_node_idx or drug2_name not in drug_name_to_node_idx:
print(f"Skipping pair ({drug1_name}, {drug2_name}) in batch {i+1} due to missing node index.")
continue
try:
# 模型 forward 需要药物名称和初始嵌入
output = model(drug1_name, drug2_name, initial_node_embeddings, drug_name_to_node_idx)
batch_outputs.append(output)
valid_indices.append(j)
except Exception as e:
print(f"Error during forward pass for pair ({drug1_name}, {drug2_name}) in batch {i+1}: {e}")
import traceback
traceback.print_exc()
continue # 跳过这个样本
# 如果批次中没有有效输出,则跳过
if not batch_outputs:
print(f"Skipping batch {i+1} due to no valid outputs.")
continue
# 将有效输出和标签组合
outputs_tensor = torch.stack(batch_outputs) # (num_valid_samples)
valid_labels = labels[valid_indices] # (num_valid_samples)
# 计算损失
loss = criterion(outputs_tensor, valid_labels)
# 反向传播和优化
loss.backward()
# 可选:梯度裁剪
# torch.nn.utils.clip_grad_norm_(model.parameters(), max_norm=1.0)
optimizer.step()
train_loss += loss.item() * len(valid_indices) # 按样本数加权损失
# 计算准确率
predictions = (outputs_tensor >= 0.5).float()
train_correct += (predictions == valid_labels).sum().item()
train_samples += len(valid_indices)
if (i + 1) % 10 == 0: # 每 10 个批次打印一次进度
print(f" Batch {i+1}/{len(train_loader)}, Loss: {loss.item():.4f}")
avg_train_loss = train_loss / max(1, train_samples)
train_acc = train_correct / max(1, train_samples)
print(f"Epoch {epoch+1} Train Summary: Avg Loss: {avg_train_loss:.4f}, Accuracy: {train_acc:.4f}")
# --- 验证阶段 ---
model.eval()
val_loss = 0
val_correct = 0
val_samples = 0
print(f"Epoch {epoch+1}/{epochs} - Validation...")
# 验证时也使用相同的初始嵌入
# initial_node_embeddings, drug_name_to_node_idx 已在上文计算
with torch.no_grad():
for i, batch in enumerate(val_loader):
if batch is None:
print(f"Skipping empty validation batch {i+1}")
continue
drug1_names = batch['drug1_name']
drug2_names = batch['drug2_name']
labels = batch['label'].to(device)
batch_outputs = []
valid_indices = []
for j in range(len(drug1_names)):
drug1_name = drug1_names[j]
drug2_name = drug2_names[j]
if drug1_name not in drug_name_to_node_idx or drug2_name not in drug_name_to_node_idx:
continue
try:
output = model(drug1_name, drug2_name, initial_node_embeddings, drug_name_to_node_idx)
batch_outputs.append(output)
valid_indices.append(j)
except Exception as e:
print(f"Error during validation forward pass for pair ({drug1_name}, {drug2_name}) in batch {i+1}: {e}")
continue
if not batch_outputs:
continue
outputs_tensor = torch.stack(batch_outputs)
valid_labels = labels[valid_indices]
loss = criterion(outputs_tensor, valid_labels)
val_loss += loss.item() * len(valid_indices)
predictions = (outputs_tensor >= 0.5).float()
val_correct += (predictions == valid_labels).sum().item()
val_samples += len(valid_indices)
avg_val_loss = val_loss / max(1, val_samples)
val_acc = val_correct / max(1, val_samples)
print(f"Epoch {epoch+1} Validation Summary: Avg Loss: {avg_val_loss:.4f}, Accuracy: {val_acc:.4f}")
print("Training finished.")
return model
准备数据并开始训练。 在开始训练之前,需要实例化 DDIDataset,提供包含药物图像路径和描述的 DataFrame、已知的药物相互作用列表、构建好的知识图谱(medical_kg)以及节点名称到索引的映射(node_to_idx)。然后,将完整的数据集划分为训练集和验证集,通常采用 80/20 的比例。接着,为训练集和验证集创建 DataLoader 实例,指定批次大小(batch size)、是否打乱数据(shuffle=True 用于训练集)以及使用前面定义的 custom_collate_fn 来处理批次数据。之后,根据知识图谱的节点类型数量和边类型数量,实例化 KGGuidedMultimodalModel。最后,调用 train_kg4mm_model 函数,传入模型、训练和验证数据加载器以及训练轮数,启动模型的训练过程。这个序列完成了从数据准备到实际模型训练的流程。
# 确保 drug_data_df 包含所需信息且与 medical_kg/node_to_idx 一致
print(f"Number of drugs with data: {len(drug_data_df)}")
print(f"Number of nodes in KG: {len(medical_kg.nodes)}")
print(f"Number of drug interactions provided: {len(drug_drug_interactions)}")
# 初始化数据集
ddi_dataset = DDIDataset(drug_data_df, drug_drug_interactions, medical_kg, node_to_idx)
print(f"Total samples in dataset: {len(ddi_dataset)}")
# 检查数据集是否为空
if len(ddi_dataset) == 0:
print("Error: Dataset is empty. Check data loading and filtering steps.")
# 可以在这里停止执行或抛出错误
raise ValueError("Dataset creation failed, resulting in an empty dataset.")
# 将数据集拆分为训练集和验证集
train_size = int(0.8 * len(ddi_dataset))
# 确保 val_size 不为负数
val_size = max(0, len(ddi_dataset) - train_size)
# 处理数据集大小不足的情况
if train_size == 0 or val_size == 0:
print("Warning: Dataset size is too small for an 80/20 split. Adjusting split or using the whole dataset for training.")
# 可以选择不同的策略,例如,如果 val_size 为 0,则不进行验证
# 这里简单地将所有数据用于训练,如果验证集大小为0
if val_size == 0 and train_size > 0:
train_dataset = ddi_dataset
val_dataset = None # 或者创建一个空的 Dataset
print("Using the entire dataset for training as validation set size is zero.")
elif train_size > 0 and val_size > 0:
train_dataset, val_dataset = torch.utils.data.random_split(ddi_dataset, [train_size, val_size])
else:
raise ValueError("Both train and validation set sizes are zero. Cannot proceed.")
else:
train_dataset, val_dataset = torch.utils.data.random_split(ddi_dataset, [train_size, val_size])
print(f"Train dataset size: {len(train_dataset)}")
if val_dataset:
print(f"Validation dataset size: {len(val_dataset)}")
else:
print("Validation dataset size: 0")
# 创建数据加载器
# 调整 batch_size 根据内存情况
batch_size = 4 # 减小 batch_size 尝试
train_loader = DataLoader(train_dataset, batch_size=batch_size, shuffle=True, collate_fn=custom_collate_fn)
# 仅当 val_dataset 有效时创建 val_loader
val_loader = DataLoader(val_dataset, batch_size=batch_size, shuffle=False, collate_fn=custom_collate_fn) if val_dataset else None
# 使用 PyG 图初始化模型
num_node_types = pyg_graph.x.shape[1] # 从 PyG Data 对象获取节点特征维度 (类型数量)
num_edge_types = len(edge_type_to_idx) # 边的类型数量
# 初始化 KG 引导的多模态模型
model = KGGuidedMultimodalModel(pyg_graph, num_node_types, num_edge_types, node_to_idx, idx_to_node)
# 训练模型 (仅当有验证加载器时传递它)
trained_model = train_kg4mm_model(model, train_loader, val_loader, drug_data_df, epochs=5) # 减少 epoch 数量进行快速测试
推理和解释。 在进行 DDI 预测时,模型首先接收待预测的两种药物的名称。系统根据名称查找对应的预处理图像和文本描述,并获取它们在知识图谱中的节点表示。训练好的模型(设置为评估模式 model.eval())接收这些输入,并结合预先计算好的初始节点嵌入和图谱结构,通过前向传播计算出一个概率分数,量化这两种药物发生相互作用的风险。同时,调用 retrieve_knowledge_subgraph 函数提取与该药物对相关的知识子图,包括直接连接、共享的生物靶点(蛋白质、疾病等)以及连接它们的间接路径。基于预测的概率分数,可以将其映射为风险等级(如低、中、高)。解释模块则整合概率分数和提取的知识子图信息,生成自然语言解释。解释内容会突出显示图谱中支持预测的关键结构,例如已知的相互作用机制(来自直接连接的边属性)、共享靶点(共同邻居)以及模型可能依赖的关键路径。最后,可以根据风险等级和图谱证据,提供示例性的临床建议或关注点,从而清晰地展示知识图谱如何指导预测过程并提供可解释的依据。
def predict_interaction(model, drug1_name, drug2_name, drug_data_df, medical_kg):
device = next(model.parameters()).device # 获取模型所在的设备
model.eval()
# 检查药物是否存在于数据和 KG 中
if drug1_name not in model.node_to_idx or drug2_name not in model.node_to_idx:
print(f"Error: {drug1_name} or {drug2_name} not found in the knowledge graph node index.")
return 0.0, retrieve_knowledge_subgraph(medical_kg, drug1_name, drug2_name) # 仍然尝试获取知识
if drug1_name not in drug_data_df['name'].values or drug2_name not in drug_data_df['name'].values:
print(f"Error: {drug1_name} or {drug2_name} not found in the provided drug data (missing image/text).")
# 可以选择返回错误或默认值
return 0.0, retrieve_knowledge_subgraph(medical_kg, drug1_name, drug2_name)
# 获取药物对的知识子图 (在预测前或后皆可)
knowledge = retrieve_knowledge_subgraph(medical_kg, drug1_name, drug2_name)
# 进行预测
with torch.no_grad():
# 推理时也需要初始节点嵌入
initial_node_embeddings, drug_name_to_node_idx = model.get_initial_node_embeddings(drug_data_df)
initial_node_embeddings = initial_node_embeddings.to(device)
# 检查推理时药物是否在 name_to_node_idx 映射中 (get_initial_node_embeddings 创建的)
if drug1_name not in drug_name_to_node_idx or drug2_name not in drug_name_to_node_idx:
print(f"Error: Node index mapping missing for {drug1_name} or {drug2_name} during inference.")
return 0.0, knowledge # 返回默认概率
interaction_prob = model(
drug1_name,
drug2_name,
initial_node_embeddings,
drug_name_to_node_idx
)
# 确保返回的是 Python float 类型
return interaction_prob.item(), knowledge
def explain_interaction_prediction(drug1_name, drug2_name, probability, knowledge):
explanation = f"KG-Guided Multimodal Analysis for Interaction between {drug1_name} and {drug2_name}:\n\n"
# 解释概率
if probability > 0.8:
risk_level = "High"
elif probability > 0.5:
risk_level = "Moderate"
else:
risk_level = "Low"
explanation += f"Predicted Interaction Risk Level: {risk_level} (Probability: {probability:.2f})\n\n"
# 基于知识图谱结构进行解释
explanation += "Knowledge Graph Analysis:\n"
has_kg_evidence = False
if knowledge.get('direct_interaction'):
mechanism = knowledge['direct_interaction'].get('mechanism', 'an unknown mechanism')
relation_type = knowledge['direct_interaction'].get('relation', 'interacts_with')
explanation += f"✓ Direct Connection: The knowledge graph indicates a direct '{relation_type}' relation between these drugs, potentially involving {mechanism}.\n"
has_kg_evidence = True
if knowledge.get('common_targets'):
explanation += f"✓ Common Neighbors: These drugs share connections with the following entities in the knowledge graph:\n"
for target in knowledge['common_targets'][:5]: # 显示前 5 个
explanation += f" - {target}\n"
if len(knowledge['common_targets']) > 5:
explanation += " - ... (and possibly others)\n"
explanation += " This suggests potential interaction through shared pathways or targets.\n"
has_kg_evidence = True
if knowledge.get('paths') and len(knowledge['paths']) > 0:
explanation += f"✓ Connecting Paths: The model identified connecting paths (up to length {3}) in the graph:\n" # 假设 max_path_length=3
for i, path in enumerate(knowledge['paths'][:3]): # 显示前 3 条路径
path_str = " → ".join(path)
explanation += f" - Path {i+1}: {path_str}\n"
explanation += " These paths highlight indirect relationships that might contribute to interactions.\n"
has_kg_evidence = True
if not has_kg_evidence:
explanation += " - No direct interaction, common neighbors, or short connecting paths found in the analyzed subgraph.\n"
explanation += "\n"
# 关注 KG 结构如何指导解释
explanation += "Multimodal Integration Insights:\n"
explanation += " - The knowledge graph structure likely guided the model to focus on specific molecular features (from images) and textual descriptions relevant to the identified graph patterns (or lack thereof).\n"
explanation += " - Graph neural networks propagated information across the graph, allowing the final prediction to be informed by the broader network context of both drugs.\n"
# explanation += " - Attention mechanisms (if used explicitly for explanation) would highlight which graph connections or multimodal features were most influential.\n\n" # 如果有注意力权重解释则添加
# 临床意义(示例 - 真实系统需要更严谨的医学建议)
explanation += "Potential Clinical Considerations (Illustrative):\n"
if risk_level == "High":
explanation += " - High predicted risk suggests careful consideration. Potential need for dose adjustment, enhanced monitoring, or alternative therapy, especially if supported by strong KG evidence (e.g., direct interaction with known mechanism).\n"
elif risk_level == "Moderate":
explanation += " - Moderate risk warrants caution. Monitoring for potential adverse effects is advisable. The specific KG findings (e.g., common targets) might guide what to monitor.\n"
else: # Low risk
explanation += " - Low predicted risk suggests standard monitoring is likely sufficient. The lack of strong KG structural links supports a lower likelihood of significant interaction.\n"
explanation += " - Always consult official drug interaction resources and clinical guidelines.\n"
return explanation
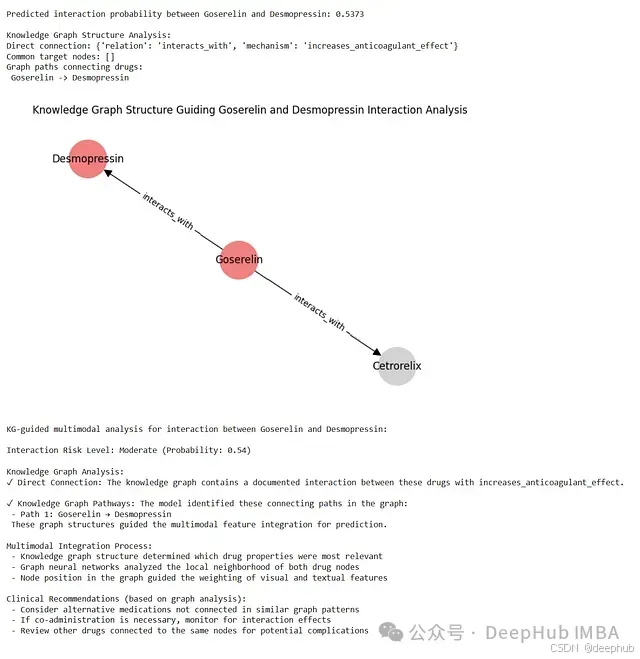
结果。 为了演示完整的工作流程,选择一对药物(例如,“Goserelin” 和 “Desmopressin”)进行 DDI 预测。系统加载这两种药物预处理后的图像和文本数据。然后,将这些多模态输入以及药物名称传递给训练好的 trained_model(处于评估模式)。模型输出一个概率分数,表示预测的相互作用风险。同时,系统调用 retrieve_knowledge_subgraph 提取与这对药物相关的知识图谱子图,包括直接连接、共享邻居以及连接路径。
接下来,可以对提取的子图进行可视化。使用 NetworkX 和 Matplotlib,绘制子图的节点和边。为了增强可读性,可以为不同类型的节点(如目标药物、蛋白质、疾病)分配不同的颜色,并标注边的关系类型。这种可视化直观地展示了支持模型预测的图谱结构。最后,调用 explain_interaction_prediction 函数,结合预测概率和提取的知识子图,生成一段自然语言解释。这段解释将概率分数与具体的图谱特征(如直接相互作用的机制、共享靶点、关键路径)联系起来,并可能包含基于风险等级和图谱证据的示例性临床建议。风险评估、可视化子图和叙述性解释共同阐明了知识图谱的拓扑结构如何指导多模态信号的融合,并为模型的预测提供了透明的依据。
# 示例用法
# drug_pair = ("Goserelin", "Desmopressin") # 示例对 1 (已知相互作用)
drug_pair = ("Lisinopril", "Albumin") # 示例对 2 (药物-蛋白质,非 DDI,预期低概率)
# drug_pair = ("Fluoxetine", "Metoprolol") # 示例对 3 (潜在相互作用,可能通过 CYP P450)
# 确保模型已训练或加载
if 'trained_model' not in locals():
print("Error: Model is not trained or loaded. Please run the training cell first.")
# 可以尝试加载已保存的模型
# model_path = "kg4mm_model.pth"
# if os.path.exists(model_path):
# model.load_state_dict(torch.load(model_path))
# trained_model = model
# else:
# raise RuntimeError("Trained model not available.")
# 临时处理:如果模型未训练,则退出或使用未训练的模型(结果无意义)
trained_model = model # 使用未训练的模型进行演示,结果不可靠
print(f"\nPredicting interaction between: {drug_pair[0]} and {drug_pair[1]}")
prob, knowledge = predict_interaction(trained_model, drug_pair[0], drug_pair[1], drug_data_df, medical_kg)
print(f"\nPredicted interaction probability: {prob:.4f}")
print("\nKnowledge Graph Structure Analysis:")
print(f"- Direct connection: {knowledge.get('direct_interaction', 'None')}")
print(f"- Common target nodes: {knowledge.get('common_targets', 'None')}")
print(f"- Graph paths connecting drugs (showing max 3):")
if knowledge.get('paths'):
for i, path in enumerate(knowledge['paths'][:3]):
print(f" - Path {i+1}: {' -> '.join(path)}")
else:
print(" None found.")
# 可视化这些药物的子图以显示 KG 引导的方法
plt.figure(figsize=(14, 9)) # 调整图形大小
subgraph_nodes = set([drug_pair[0], drug_pair[1]])
# 添加知识子图中的节点到可视化范围
if knowledge.get('direct_interaction'):
# 如果有直接边,确保两个节点都在
pass # 它们已经是 subgraph_nodes 的成员
if knowledge.get('common_targets'):
subgraph_nodes.update(knowledge['common_targets'])
if knowledge.get('paths'):
for path in knowledge['paths']:
subgraph_nodes.update(path)
# 添加一层邻居以显示 KG 中的上下文 (可选,可能使图变复杂)
# neighbors_to_add = set()
# for node in list(subgraph_nodes): # 使用列表副本迭代,因为集合在循环中可能被修改
# if node in medical_kg:
# # 限制邻居数量避免图形过于拥挤
# neighbors_to_add.update(list(medical_kg.neighbors(node))[:2])
# neighbors_to_add.update(list(medical_kg.predecessors(node))[:2]) # 也考虑前驱节点
# subgraph_nodes.update(neighbors_to_add)
# 确保所有节点都在 medical_kg 中
valid_subgraph_nodes = [n for n in subgraph_nodes if n in medical_kg]
subgraph = medical_kg.subgraph(valid_subgraph_nodes)
# 检查子图是否为空
if not subgraph.nodes():
print("\nSubgraph is empty, cannot visualize.")
else:
# 使用不同的颜色表示节点类型以强调 KG 结构
node_colors = []
for node in subgraph.nodes():
node_type = subgraph.nodes[node].get('type', 'unknown')
if node == drug_pair[0] or node == drug_pair[1]:
node_colors.append('salmon') # 目标药物颜色
elif node_type == 'protein':
node_colors.append('skyblue')
elif node_type == 'disease':
node_colors.append('lightgreen')
elif node_type == 'drug':
node_colors.append('lightcoral') # 其他药物
else:
node_colors.append('lightgray') # 其他类型或未知
# 布局算法
try:
# spring_layout 对于较多节点可能效果不佳,可以尝试其他布局
pos = nx.spring_layout(subgraph, k=0.5, iterations=50, seed=42) # 调整参数 k
# pos = nx.kamada_kawai_layout(subgraph) # 另一种布局
except nx.NetworkXException as e:
print(f"Layout calculation failed: {e}. Using random layout.")
pos = nx.random_layout(subgraph, seed=42)
nx.draw(subgraph, pos, with_labels=True, node_color=node_colors,
node_size=2500, font_size=9, arrows=True, arrowsize=15, edge_color='gray')
# 标注边关系 (如果存在)
edge_labels = {}
for u, v, data in subgraph.edges(data=True):
if 'relation' in data:
edge_labels[(u, v)] = data['relation']
nx.draw_networkx_edge_labels(subgraph, pos, edge_labels=edge_labels, font_size=8)
plt.title(f"Knowledge Graph Subgraph for {drug_pair[0]} and {drug_pair[1]}", size=14)
# 保存图像前确保目录存在
os.makedirs("results", exist_ok=True)
plt.savefig(f'results/kg_subgraph_{drug_pair[0]}_{drug_pair[1]}.png')
plt.show()
# 显示解释
explanation = explain_interaction_prediction(drug_pair[0], drug_pair[1], prob, knowledge)
print("\n--- Interaction Explanation ---")
print(explanation)
以 Goserelin 和 Desmopressin 为例进行测试时,(假设训练后的)模型可能返回一个概率值,例如 0.54,这可以被解释为中等相互作用风险。知识图谱分析显示,存在一个从 Goserelin 指向 Desmopressin 的直接 interacts_with 边,其 mechanism 属性为 increases_anticoagulant_effect。假设在此示例中未找到共享的蛋白质或疾病节点,模型的主要依据便是这个直接的、有记录的相互作用机制。在子图可视化中,Goserelin 和 Desmopressin 节点会以特定颜色(如红色或鲑鱼色)突出显示,连接它们的有向边清晰可见,直观地展示了驱动预测的关键关系。
总结
KG4MM 方法通过将知识图谱置于多模态学习流程的核心,展示了其在融合分子图像和文本描述等异构数据方面的潜力,其效果优于仅依赖单一数据源的方法。该框架下的每个预测都能够得到知识图谱中显式证据的支持——无论是直接的边、共享的靶点节点还是连接路径——从而将预测结果与潜在的生物学或化学机制联系起来。通过这种方式,KG4MM 不仅有望提升在生物化学、材料科学、医学诊断等领域中预测任务的性能,而且其内在的结构化知识表示也为模型提供了更强的可解释性。
参考文献:
Knowledge Graphs Meet Multi-Modal Learning: A Comprehensive Survey. arXiv:2402.05391.

















 1239
1239

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









