






摘要
变分量子特征求解器 (VQE) 的研究最近备受关注,因为它们可能导致近期量子设备的实际应用。然而,它们的性能取决于所用变分假设的结构,这需要平衡相应电路的深度和表达能力。近年来,已经引入了各种 VQE 结构优化方法,但机器学习在帮助解决这一问题方面的能力尚未得到充分研究。在这项工作中,我们提出了一种强化学习算法,该算法可以自主探索可能的假设空间,识别仍能产生准确基态能量估计的经济电路。该算法具有内在动机,它在最小化电路深度的同时逐步提高结果的准确性。我们展示了我们的算法在估计氢化锂 (LiH) 基态能量问题上的性能。在这个众所周知的基准问题中,我们实现了化学准确性,以及电路深度方面最先进的结果。
方法
量子线路的在(NISQ)技术背景下,为了最大化限度地减少门误差和退相干的不利影响,量子线路的设计原则是尽可能设计的浅,尽可能节省门。在分子预测中,计算化学系统的基态能量和低能特性(化学结构问题)。
目标是准备状态,该状态可以通过变分原理来近似给定哈密顿量H的基态。
众所周知,假设的结构可以显著的影响VQE的性能,因为估计的基态与真实基态的接近程度取决于假设可访问的状态的流行。因此,找到新的架构构建的方法可能会导致化学量子变分方法的突破。
例如,对于强关联系统,标准假设可能不适用。最重要的假设主要分为两类:受到化学启发的(酉耦合簇假设和受到硬件启发的硬件高效假设)。这两类假设众所周知,假设的结构可以显著影响 VQE 的性能 ,因为估计的基态与真实基态的接近程度取决于假设可访问的状态流形。因此,找到新的架构构建方法可能会导致化学量子变分方法的突破(例如,对于强关联系统,标准假设可能不适用),但也可能导致机器学习和优化等利用变分电路的其他领域的突破 。)。这两类架构都需要使用固定架构来确定酉 U (θ),从而确定相应的假设。然后,通常会将模拟电路分解为两量子位 CNOT 和一量子位旋转门,参数为 (θ1, θ2, . . . , θn) ∈ [0, 2π],并通过经典子程序进行优化。但是,架构本身也可以进行优化。这会导致结构优化问题变得困难,因为它是一个组合优化问题,必须平衡两个相互竞争的因素。一方面,模拟电路需要具有足够的表达能力,以保证对基态能量的良好近似。另一方面,需要控制电路的深度和大小,以便后者与 NISQ 限制兼容。
强化学习问题的量子门优化
整个环境的动态由两个运算符控制,它们负责在给定当前状态和动作和一个奖励函数
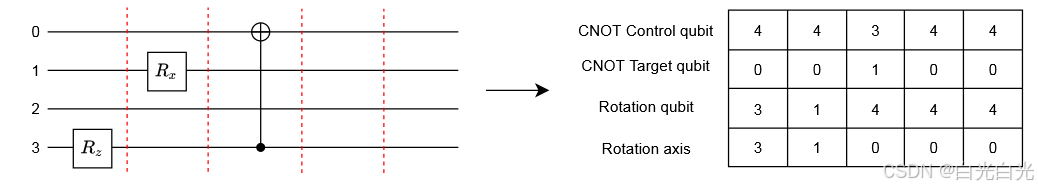
为了利用深度强化学习算法,我们以神经网络友好的形式设计了状态和动作的表示。即,每个状态都表示为由单个量子门组成的有序层列表。该列表唯一地定义了整个量子电路。因此,列表中元素的顺序描述了电路中量子门的顺序。为了构建电路,我们使用 CNOT 和一个量子比特旋转门 - 它们可以在当前可用的量子设备上实现。CNOT 门由两个值编码,表示控制和目标量子位的位置(位置从 0 开始枚举)。这样,代理具有完全的灵活性,原则上可以在两个任意量子位之间插入 CNOT 门(相同量子位除外)。旋转门用相应的位置号(从 0 开始)编码,表示要应用旋转的量子位和旋转轴(即产生旋转的 Pauli 算子 X、Y、Z,它们表示为整数 1、2、3)。为了完整描述旋转门,我们需要指定旋转角度,但是我们故意从状态表示中省略了这个参数。在每个状态表示的末尾,我们附加了该状态的估计能量。在我们的方法中,用户需要定义电路层的最大数量 (L)。如果给定电路的层数少于 L,则剩余的层数将用恒等运算符填充(参见图 1)。
![]()
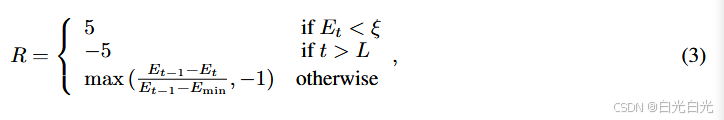
其中 ξ 是预定义的阈值,表示想要接近准确能量的程度。关于 ξ 的选择的更多细节将在第 3.3 节中讨论。因此,如果能量低于阈值,则给出最大奖励 +5,而如果电路有超过 L 层,则给出最小奖励 −5。如果电路既不满足阈值 ξ 也未超过最大层数 L,则给出中间奖励。每当时间步 t 的估计值明显差于 t − 1 的估计值时,这个中间奖励(理想情况下等于 1)的上限为 −1(回想一下
≥ 0),极限情况下是+5或者-5,当
,即是加入中间项,避免高度稀疏的奖励。
代理和环境的规范
在本文中,我们采用了具有 greedy 策略和 ADAM 优化器的双深度 Q 网络 (DDQN)。DDQN 是一种 RL 方法,类似于标准深度 Q 网络 ,它从神经网络近似的 Q 函数中得出策略。然而,DDQN 通过使用两个独立的网络来减少高估偏差,一个用于 Q 值评估,另一个用于选择动作,前者是后者的早期副本。根据等式 2,为了估计特定电路获得的能量,需要确定连续值旋转门角度。我们使用完善/标准的连续优化方法,例如线性近似约束优化 (COBYLA) 和 Rotosolve。虽然我们也可以为此目的使用代理,但我们出于两个主要原因不这样做。首先,在这项工作中,我们旨在评估 RL 对电路结构优化的有效性,而不是参数搜索。后者在其他文献中得到了广泛的研究,因此被视为一个单独的子程序,可以通过其他更发达的工具来解决。第二个原因是实际的,因为训练代理可靠地预测角度值将导致训练时间明显延长。出于上述原因,我们的方法是混合的:代理学习如何在给定由独立优化算法确定的一组参数的情况下构建特定电路并获得奖励。我们仅在代理将旋转门附加到电路的步骤之后应用角度优化子程序,从而无需在状态表示中包含参数。在给定步骤中优化的角度数量是我们方法中的超参数,其选择将在第 4.2 节中分析。在这项工作中,我们考虑优化所有一次优化多个角度(全局策略),以及一次优化几个角度(局部策略)。这些不同的实验设置使我们能够检查仅优化电路角度子集所获得的粗略能量近似是否足以创建足够好的电路。
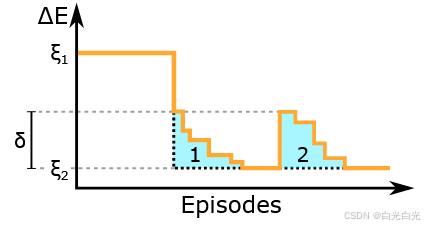
内在动机和课程学习模型
参考具体论文:
Curriculum learning | Proceedings of the 26th Annual International Conference on Machine Learning
实验评估
表 1:比较使用阈值 RL 方法获得的最小深度和门数与使用标准 HE 和 UCCSD ansta ̈ze 获得的实现化学精度的电路的最小深度和门数。实验是在键距为 2.2 ̊A 的 6 量子比特 LiH 分子上进行的。对于 RL 数据,“avg”表示不同试验中最小深度的平均值,而“min”表示所有试验中达到的最小值。对于标准 ansta ̈ze,最小深度和门数显然由架构本身固定。RL 方法在 10 次试验中的 2 次中成功生成了实现化学精度的电路。

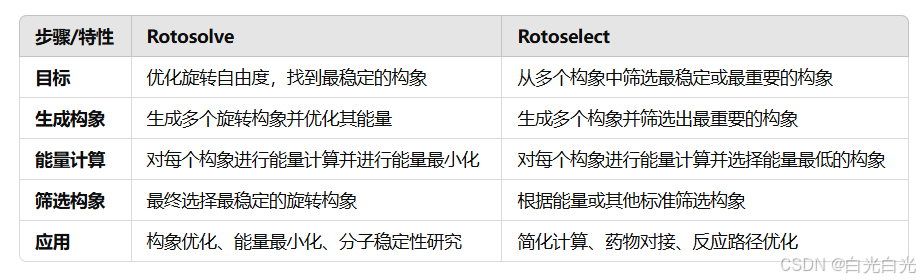
附录:1.什么是Rotosolve和Rotoselect?
Rotosolve:
-
Rotosolve 是一种在分子模拟中使用的方法或算法,用于求解和优化分子的旋转自由度,尤其是在考虑分子中某些原子或基团相对旋转时。这个过程常见于处理有多个自由旋转键的分子。
-
通常在分子动力学或量子力学计算中,分子内的一些部分可以绕某些化学键自由旋转。Rotosolve 方法帮助找出这些旋转模式与分子稳定性之间的关系,或者在某些情况下,它可以用于计算不同构象(conformers)之间的能量差异。
-
例如,在计算一个包含苯环的分子时,苯环可能在某些条件下旋转,影响分子的整体形状和能量分布。Rotosolve 算法通过求解不同旋转自由度对分子能量的影响,优化分子的结构。
-
应用:
- 研究分子旋转自由度的影响,例如在药物设计中,分子可能有多个构象,每个构象的稳定性和生物活性不同。
- 优化分子构象,尤其是在分子模拟和能量计算中。
Rotoselect:
-
Rotoselect 是指在分子模拟中选择特定的旋转自由度,或从多个可能的旋转构象中选择一个最可能的构象。这通常是在计算多个旋转构象时,从中选择最具代表性的一个或几个旋转构象进行进一步研究。
-
该过程可以通过筛选和选择最符合某些标准(如能量最低、最稳定、与实验数据最接近等)的旋转构象来减少计算负担。这有助于减少不必要的计算并集中研究最重要的构象。
-
应用:
- 分子动力学模拟:在模拟分子的动态行为时,可能需要考虑多个旋转自由度。Rotoselect 可以用来选择最重要的旋转构象,优化模拟过程。
- 分子对接(Docking):在药物设计中,Rotoselect 可以帮助选择最可能与靶标蛋白结合的构象。
- 能量最优化:通过选择最稳定的旋转构象,降低计算复杂度。
总结
- Rotosolve 主要指求解分子旋转自由度的优化过程,目的是找出旋转构象对分子能量和稳定性的影响。
- Rotoselect 是在多个旋转自由度中选择最合适的构象或旋转模式,通常用于简化计算或优化分子模拟。
Rotosolve 算法实现过程
Rotosolve 旨在通过优化分子中旋转自由度的构象,求解每个旋转构象的稳定性,通常依赖于分子能量的最小化。下面是实现过程的详细步骤:
步骤 1: 输入分子结构
- 输入是一个初始的分子结构(例如,通过化学结构式或从PDB、SMILES等格式加载的分子数据)。
- 该分子结构包含旋转自由度,如单键(C-C 单键、C-H 单键)周围的旋转轴。
步骤 2: 识别旋转自由度
- 对于每一个化学键,算法需要识别其旋转自由度。例如,在C-C键上,碳原子和氢原子之间的旋转自由度可以导致不同的构象。
- 需要确定哪些键可以旋转,哪些旋转会影响分子的几何结构。比如在环状分子中,某些键可能限制旋转自由度。
步骤 3: 构建旋转构象
- 对于每个旋转自由度,生成不同的旋转角度,这样就能产生不同的构象。每次旋转会导致新的分子几何结构。
- 例如,如果C-C键的旋转自由度可以从0°到360°变化,Rotosolve 会尝试多个角度(例如:0°, 90°, 180°, 270°等)生成多个构象。
步骤 4: 计算能量
- 对每个生成的旋转构象,计算其总能量。这个计算通常通过量子力学方法(如密度泛函理论(DFT))或经典力场方法(如AMBER、CHARMM等)进行。
- 计算能量时,要考虑到分子内部的各种相互作用,如原子间的库伦力、范德华力、键合力、角度弯曲等。
步骤 5: 能量最小化
- 对每个构象进行能量最小化。使用优化算法(例如共轭梯度法、牛顿法或分子动力学)来调整分子几何,使能量达到局部最小值。
- 通过最小化能量来找到稳定构象。这是Rotosolve的核心步骤:通过调整旋转角度来找到最稳定的构象。
步骤 6: 选择最稳定的构象
- 最终,通过能量计算和最小化,选择最稳定的构象作为输出。
- 如果有多个最低能量的构象,可能会将它们都输出,供后续分析使用。
步骤 7: 输出结果
- 输出最稳定的旋转构象和其对应的能量。这些数据可以用于进一步的分子动力学模拟、药物设计或其他应用。
Rotoselect 算法实现过程
Rotoselect 主要用来从多个旋转构象中筛选出最有代表性、最可能出现的构象,以减少计算量或聚焦在重要的构象上。实现过程的步骤如下:
步骤 1: 输入分子结构
- 与 Rotosolve 相同,输入的是一个分子结构。这个分子包含多个旋转自由度,可能会生成多个旋转构象。
步骤 2: 生成多个旋转构象
- 对分子中的每一个旋转自由度,生成多个可能的构象。例如,对于一个C-C单键,可以尝试不同的旋转角度(如0°, 90°, 180°, 270°)来生成多个不同构象。
- 如果有多个旋转自由度,构象数目将呈指数级增长。比如,两个自由度每个旋转自由度有4个角度,则会生成4 × 4 = 16种构象。
步骤 3: 计算构象能量
- 对于生成的每个构象,使用量子力学计算(如DFT)或经典力场计算来得到每个构象的总能量。
- 通过能量计算,可以了解每个构象的稳定性,能量较低的构象通常是最稳定的。
步骤 4: 筛选构象
- 根据能量、稳定性或者其他标准筛选构象。通常,会选择能量最低的构象,或者选择能量相近但具有重要空间特征的构象。
- 例如,可以设定一个能量阈值,筛选出能量低于阈值的构象,排除那些能量较高的构象。
步骤 5: 选择特定构象
- 如果有多个旋转构象具有相似的低能量,可以进一步通过其他标准(如与实验数据的匹配程度、反应性等)来筛选最终的构象。
- 例如,在药物对接中,可能选择与靶标蛋白结合最可能的构象,而不一定选择所有最低能量的构象。
步骤 6: 输出结果
- 输出选择的构象及其对应的能量。这些构象可以用于后续的动力学模拟、对接实验或者其他研究。
2.如何测试变分量子算法,并找到相关数据集,建立和测试模型?
1. 量子化学数据集
- 量子化学领域是变分量子算法(VQA)最常见的应用之一。VQA 可以用来求解量子化学问题,如分子的基态能量、电子结构和分子模拟等。
常见数据集:
- QM9 数据集:
- 内容:QM9 是一个包含约13万种有机分子的数据集,每个分子包含分子结构及其基态能量、分子振动频率、电偶极矩等信息。
- 用途:可以用于量子化学的计算,特别适合用来训练变分量子算法模型。
- 获取方式:可以从QM9 dataset网站下载。
- ANI 数据集:
- 内容:ANI 数据集包含大量分子的原子结构和能量数据,适用于使用深度学习方法和量子算法进行预测。
- 用途:这个数据集适合用于训练量子算法,帮助改进分子的能量预测。
- 获取方式:ANI-1 数据集可以在该网站找到。
注意:这些数据集通常需要结合传统的量子化学计算(如基于密度泛函理论(DFT)的计算)来生成,而变分量子算法可以用来从量子计算角度优化预测。
2. 量子机器学习数据集
变分量子算法也可以应用于量子机器学习任务,如分类、回归和聚类等。你可以使用一些标准的经典机器学习数据集,并将其转化为量子格式进行测试。
常见数据集:
- Iris 数据集:
- 内容:包含 150 个鸢尾花样本的数据集,包含 4 个特征维度(例如花瓣长度、花萼宽度等),适用于分类任务。
- 用途:可以将其转化为量子机器学习模型的输入,用来测试变分量子分类算法。
- 获取方式:可以从经典机器学习库(如Scikit-learn)中获取。
- MNIST 数据集:
- 内容:一个包含 7 万张手写数字的图像数据集,适用于图像分类任务。
- 用途:该数据集可以用于测试量子神经网络(QNN)和变分量子分类器。
- 获取方式:MNIST Dataset。
量子机器学习工具包:
- TensorFlow Quantum (TFQ) 或 PennyLane 等量子机器学习库可以帮助你将经典数据集映射到量子算法上,并进行模型训练与测试。
3. 量子优化数据集
量子优化问题是变分量子算法的另一重要应用,尤其是在组合优化、图着色、旅行商问题等领域。
常见数据集:
- QAP(Quadratic Assignment Problem) 数据集:
- 内容:这是一个经典的组合优化问题,涉及如何优化工厂布局以最小化运输成本等。
- 用途:适用于变分量子优化算法来求解优化问题。
- 获取方式:QAPLIB 提供了相关数据集。
- 旅行商问题(TSP):
- 内容:包含多个城市之间的最短路径问题。
- 用途:可以用来测试量子优化算法的表现,尤其是在求解大规模优化问题时。
- 获取方式:TSP问题的数据集通常可以从数学优化库或研究文献中找到。
4. 量子计算框架与模拟器
你还可以使用一些量子计算框架和模拟器来生成数据,进行量子算法的训练和测试。
常见量子计算框架和模拟器:
- IBM Qiskit:
- 功能:Qiskit 提供了多种量子算法和量子计算模拟工具,可以用来生成数据集并实现变分量子算法。Qiskit 包含了量子优化和量子机器学习工具。
- 获取方式:Qiskit 官网
- PennyLane:
- 功能:PennyLane 提供了量子机器学习和量子优化算法的库,可以方便地将量子算法应用到经典数据集上。
- 获取方式:PennyLane 官网
- Cirq (Google):
- 功能:Cirq 是 Google 提供的量子编程框架,支持量子计算和量子机器学习。
- 获取方式:Cirq 官网
5. 量子数据集的创建
如果没有现成的合适数据集,也可以自己创建数据集。你可以利用量子计算机模拟器来生成一些特定问题的量子数据集。常见的做法是:
- 利用量子计算模拟器(如 Qiskit 或 Cirq)来生成一些简单量子系统(如量子比特的纠缠状态或量子测量结果)的数据。
- 使用变分量子算法(VQA)来预测量子态的演化或量子门的效果,并生成这些计算结果的数据集。
6. 开源资源和论坛
- Quantum Open Source Foundation (QOSF) 提供了很多量子算法和量子应用的研究资源。
- 你还可以在 arXiv 或 Google Scholar 上查找相关的学术论文,了解领域内最新的数据集和测试方法。
小结:如何找到和使用数据集
- 量子化学:使用 QM9、ANI 等数据集,这些数据集可以帮助你研究分子的结构与能量。
- 量子机器学习:使用经典数据集如 MNIST、Iris,并将其映射到量子计算框架。
- 量子优化:TSP、QAP 等优化问题的数据集适合用于变分量子优化。
- 量子计算框架:利用 IBM Qiskit、PennyLane、Cirq 等工具生成和处理数据
参考:
1.https://zhuanlan.zhihu.com/p/670374419
2.Curriculum learning | Proceedings of the 26th Annual International Conference on Machine Learning


















 3198
3198

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











