






***********************该笔记为初学者笔记,仅供个人参考谨慎搬运代码******************************
samtools 排序压缩和 featureCounts 生成基因计数表
SAM文件和BAM文件
1.SAM格式:是一种通用的比对格式,用来存储reads到参考序列的比对信息,以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果。SAM分为两部分,注释信息和比对结果部分。
2.BAM格式:SAM的二进制文件,bam文件优点:bam文件为二进制文件,占用的磁盘空间比sam文本文件小;利用bam二进制文件的运算速度快。
#排序压缩步骤
conda info --envs
conda activate py2env
#下载subread
conda install -c bioconda subread
#测试是否成功
featureCounts -h#成功
cd /home/yinwen/biosoft/hisat2
#压缩:把 sam文件 转为 bam文件
samtools view -S genome.sam -b > genome.bam
#排序:samtools sort
samtools sort -n -@ 5 genome.bam -o genome
#计数统计:
featureCounts -T 5 -t exon -g Parent -a genome.gff -o genome.counts -p genome
#samtools详情:http://www.360doc.com/content/23/0927/14/1098188476_1098188476.shtml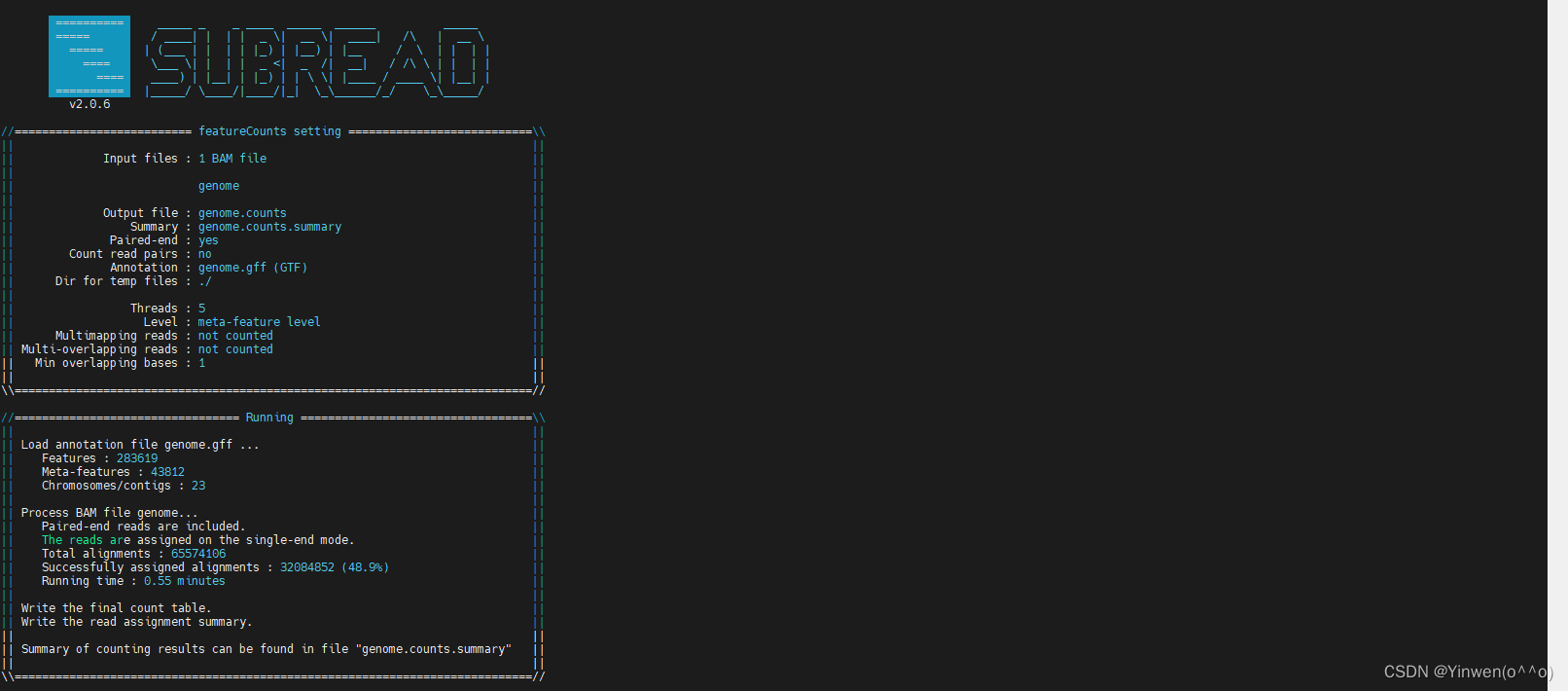
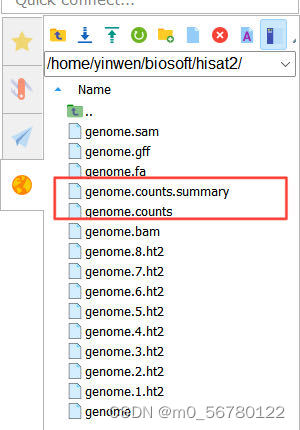
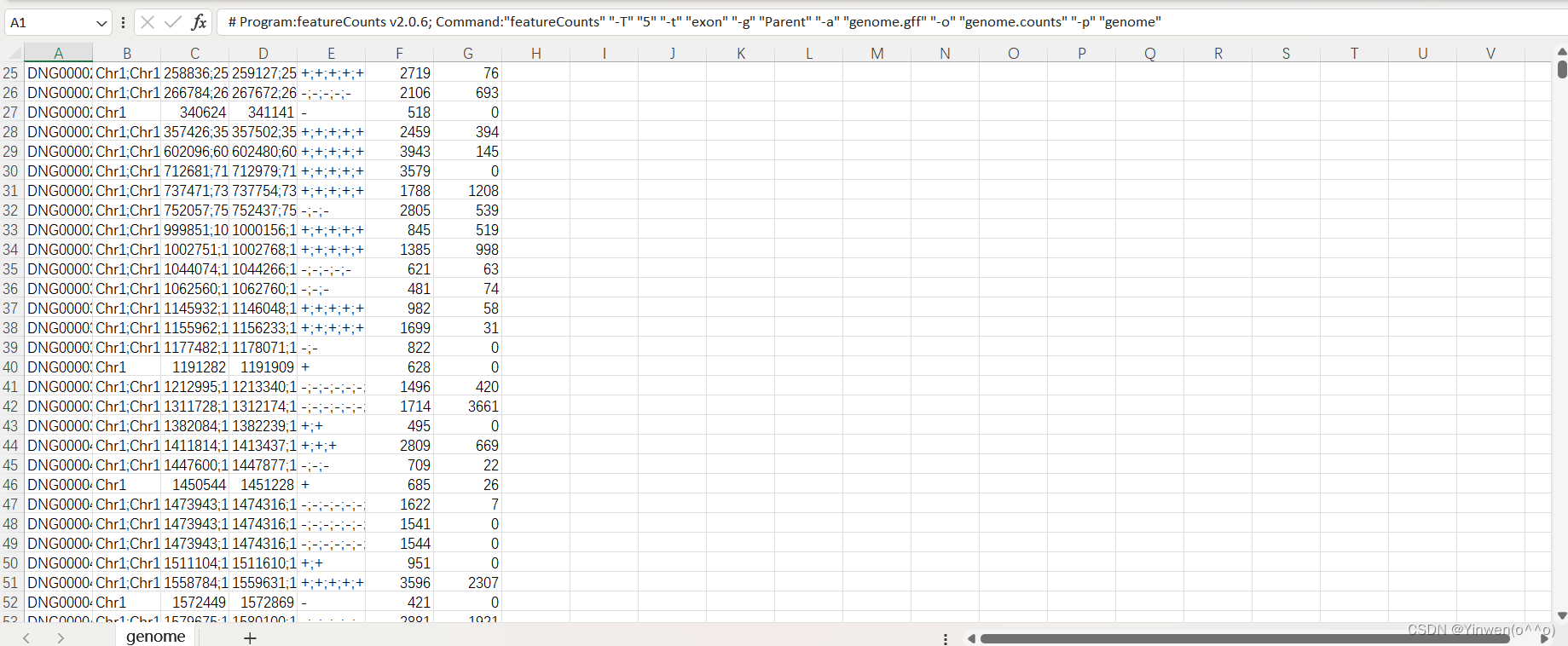
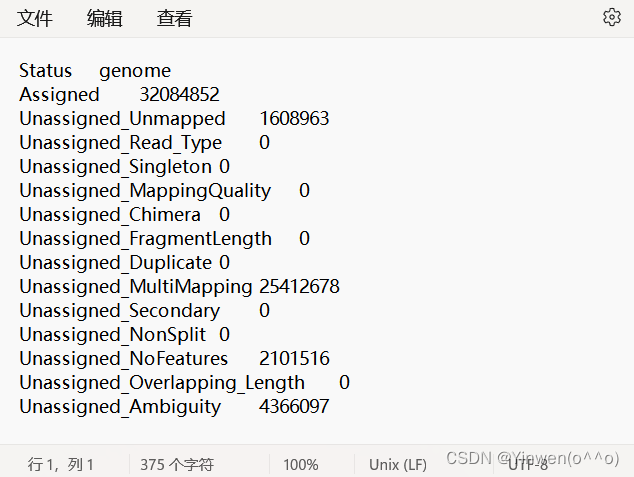
genome.gff 就是最初下载的注释文件,如果要统计多个文件的话,在-p 后面跟上就可以,会生成 genome.counts、genome.counts.summary 两个文件,我们的DG文件也是成功得到如下:















 2895
2895











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









