







下面主要讲解下 详细比对部分字段的具体含义:
alignment section部分: 如果其内容没有获得,可以用*或者0代替。
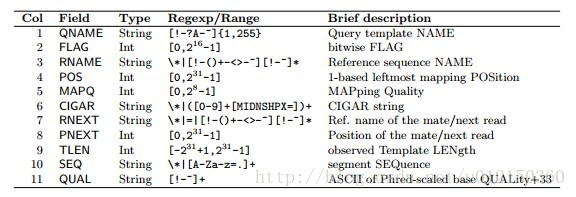
---------------------------------------------chromosome至CIGAR的信息都是非常重要的
其中:
1. QNAME 表示的是查询序列的名称即短片段(reads)的名称;
2. FLAG 以整数来表示比对的结果,不同数值有不同的意义,数值也可以是下列数的组合;

比如如果FLAG是4的话则表示该reads没有比对到参考序列上,flag为16表示single-end reads比对到参考序列的反链上,flag为83(64+16+2+1)表示paired-end reads中的第一个reads比对到参考序列上了。
3. RNAME 表示参考序列的名称,比如基因组的染色体编号等,如果没有比对上则显示为*; 【chromosome】
4. POS 表示比对的起始位置,以 1 开始计数,如果没有比对上则显示为 0 ; 【5'端起始位置】
5. MAPQ 比对质量;(数字越大,特异性越高)
6. CIGAR CIGAR 字符串,即比对的详细情况, 记录插入,删除,错配,后剪切拼接的接头。简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字+字母表示比对结果。比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一 个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
7. RNEXT 双末端测序中下一个reads比对的参考系列的名称,如果没有则用 " * " 表示,如果和前一个reads比对到同一个参考序列则用" = "表 示; 【mate名称,记录mate pair信息】
8. PNEXT 下一个reads比对到参考序列上的位置,如果没有则用 0 表示; 【 mate的位置】
9. ISIZE/TLEN query序列的模板长度或者插入长度,Template的长度, 最左边得为正+,最右边的为负 -,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,表示为 0 ;
10. SEQ reads的序列信息;
11. QUAL reads的序列质量信息,同FASTQ。
-----------后面还有些可选字段,比如:
解释:

其中可以看出Aligenment 2 和 Alignment 3是成对的reads,其插入长度为314。
bam格式中的b是binary的意思,是sam格式的二进制表示方式,为什么要用二进制表示呢? 因为sam格式文件大小通常是十分大的,一般是以G为单位,所以为了减少存储量等因素而将sam转换为二进制格式以便于分析。
sam/bam格式是由特定的一些软件(比如samtools)来处理的,包括格式互转、排序、建立索引、搜寻突变等操作,后续分析中会详细讲解samtools工具的使用方法。
【数据格式说明】
1.bam文件读取
samtools view xxx.bam
samtools view xxx.bam |less
2.bam和sam的区别与一致
(1) sam是带有比对信息的序列文件(即告诉你这个reads在染色体上的位置等),用于储存序列数据(SAM format is a generic format for storing large nucleotide sequence alignments. ) (2) BAM is the compressed binary version of the Sequence Alignment/Map (SAM) format. 生物信息中的二进制文件主要是为了节约空间,计算机机可读。可以用samtools工具实现sam和bam文件之间的转化。
二者都是fastq文件经过序列比对或者mapping后输出的格式(其储存的信息都是一致的)
【flag】
1 : 代表这个序列采用的是PE双端测序
2: 代表这个序列和参考序列完全匹配,没有错配和插入缺失
4: 代表这个序列没有mapping到参考序列上
8: 代表这个序列的另一端序列没有比对到参考序列上,比如这条序列是R1,它对应的R2端序列没有比对到参考序列上
16:代表这个序列比对到参考序列的负链上
32 :代表这个序列对应的另一端序列比对到参考序列的负链上
64 : 代表这个序列是R1端序列, read1;
128 : 代表这个序列是R2端序列,read2;
256: 代表这个序列不是主要的比对,一条序列可能比对到参考序列的多个位置,只有一个是首要的比对位置,其他都是次要的
512: 代表这个序列在QC时失败了,被过滤不掉了(# 这个标签不常用)
1024: 代表这个序列是PCR重复序列(#这个标签不常用)
2048: 代表这个序列是补充的比对(#这个标签具体什么意思,没搞清楚,但是不常用)
上面的这几个标签都是2的n次方,这样的数列有一个特点,就是随机挑选其中的几个,它们的和是唯一的,比如65 只能是1 和 64 组成,代表这个序列是双端测序,而且是read1
samtools 中flag 可以查看flags详细信息:如:
$samtools flags 770x4d 77 PAIRED,UNMAP,MUNMAP,READ1
flags值为77
PAIRED表示这条序列采用双端测序, 其值为1;
UNMAP表示这个序列没有mapping到参考序列上, 其值为4;
MUNMAP表示这个序列的另一端序列没有比对到参考序列上, 其值为8;
READ1表示这条序列是R1端序列,其值为64.
以上数值相加和为77
$samtools flags 141
0x8d 141 PAIRED,UNMAP,MUNMAP,READ2
flags值为141
PAIRED表示这条序列采用双端测序, 其值为1;
UNMAP表示这个序列没有mapping到参考序列上, 其值为4;
MUNMAP表示这个序列的另一端序列没有比对到参考序列上, 其值为8;
READ1表示这条序列是R1端序列,其值为128.
以上数值相加和为141
【samtools】
http://genome.sph.umich.edu/wiki/SAM ,
https://samtools.github.io/hts-specs/SAMv1.pdf
http://www.cnblogs.com/xudongliang/p/5437850.html,
https://genome.ucsc.edu/goldenPath/help/bam.html【****】
阿飞与安达:http://liuwei441005.blog.163.com/blog/static/135705811201322331740144/















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









