






RNA-seq:5-sratoolkit 下载数据
#下载 sratoolkit
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.0.7/sratoolkit.3.0.7-centos_linux64.tar.gz
#解压安装包
tar xvf sratoolkit.3.0.7-centos_linux64.tar.gz
#检查安装成功
cd /home/yinwen/biosoft/sratoolkit.3.0.7-centos_linux64/bin #看看能不能打开bin
#配置
vim ~/.bashrc#编辑
export PATH="/home/yinwen/biosoft/sratoolkit.3.0.7-linux64/bin:$PATH"
source ~/.bashrc#重启drwxrwxr-x 5 yinwen yinwen 156 Aug 22 10:49 sratoolkit.3.0.7-centos_linux64
-rw-rw-r-- 1 yinwen yinwen 93172877 Aug 22 10:49 sratoolkit.3.0.7-centos_linux64.tar.gz
#创造新的文件夹project/写入id
mkdir project
cd project
mkdir airway
cd airway
cat > id
SRR1039508
SRR1039509
SRR1039510
SRR1039511
SRR1039512
SRR1039513
SRR1039514
SRR1039515
SRR1039516
SRR1039517
SRR1039518
SRR1039519
SRR1039520
SRR1039521
SRR1039522
SRR1039523
cat id#构建循环下载样本;prefetch + 样本 ;挂后台
cat id |while read id ;do (prefetch $id &);done
#下载的慢,但是安全
#下载的内容为sra版本,将后缀为.sra转移到其他文件夹中
#数据大小都正常,但是SRR1039513下载失败了
#下载fastqc将sra转为fastq
#一般主要是处理fastq文件,很少转化,手里没有数据除外wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.12.1.zip
unzip fastqc_v0.12.1.zip
#配置环境变量
echo 'export PATH=~/Biosofts/FastQC:$PATH'>>~/.bashrc
source ~/.bashrc#直接转为fastq格式
fastq-dump --gzip --split-3 -O ./ /home/yinwen/project/sra/SRR1039508.sra
#解压一下.gz
gzip -d SRR1039508_1.fastq.gz
head SRR1039508_1.fastq
RNA-seq:6-qc-1
#用于分析高通量测序数据的质量,包括质量分数、测序错误、测序片段的长度分度等
#其中[options]代表选项参数,filename代表是要处理文件的名称
#分析完成后,会生成一个质量报告文件,以.HTML储存
fastqc + [options] +filename#示例
fastqc SRR1039508_2.fastq.gz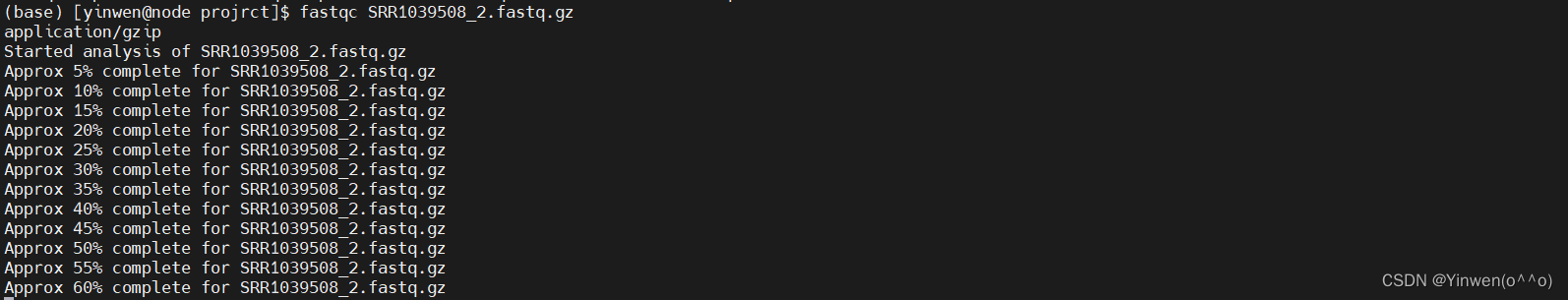
![]()
# 创造conda环境
conda info --envs #查看环境
conda create -n 环境名称 #创造环境
conda activate 环境名称 #激活环境
conda deactivate #关闭环境
conda list #查看下载的软件
#检查是否安装java版本
which java
java -version
#利用conda安装fastqc
conda install -c bioconda fastqc#文件异常关闭,编辑器出不去
rm -f .bashrc.swp# 然后跑不出来报告,一直显示java报错,还把miniconda给卸了,bin环境也让我搞得乱七八糟......
#解决方法如下
#建立python2.7的环境,大部分的转录组信息都需要在Python2的环境下进行
conda create -n py2env python=2.7
source activate py2env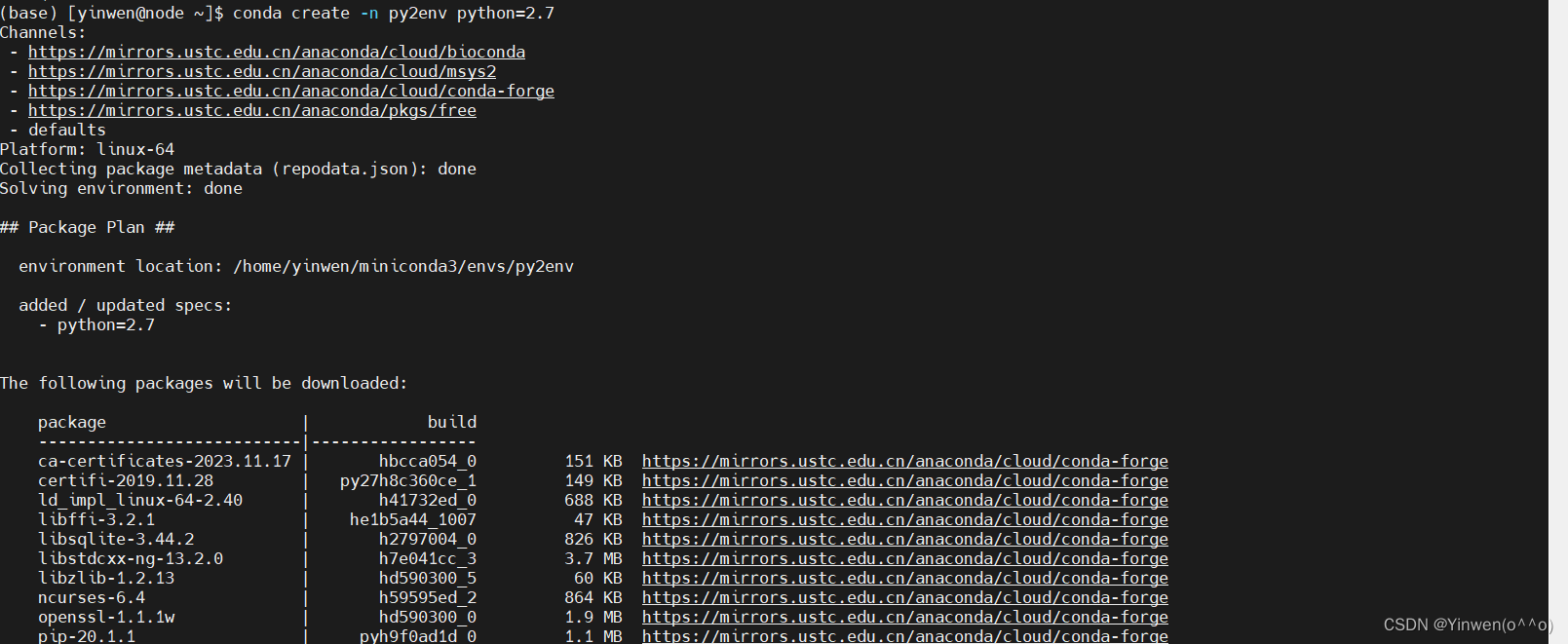
#查看一下我下载的fastqc的版本号以及帮助信息栏,看看fastqc是不是下载好的
fastqc --version
fastqc -h(py2env) [yinwen@node ~]$ fastqc --version
FastQC v0.12.1#然后开始跑报告就ok,先跑了一个,成功
fastqc -t 15 /home/yinwen/biosoft/DNG_part/DG5_1_R1.fq.gz -o ~/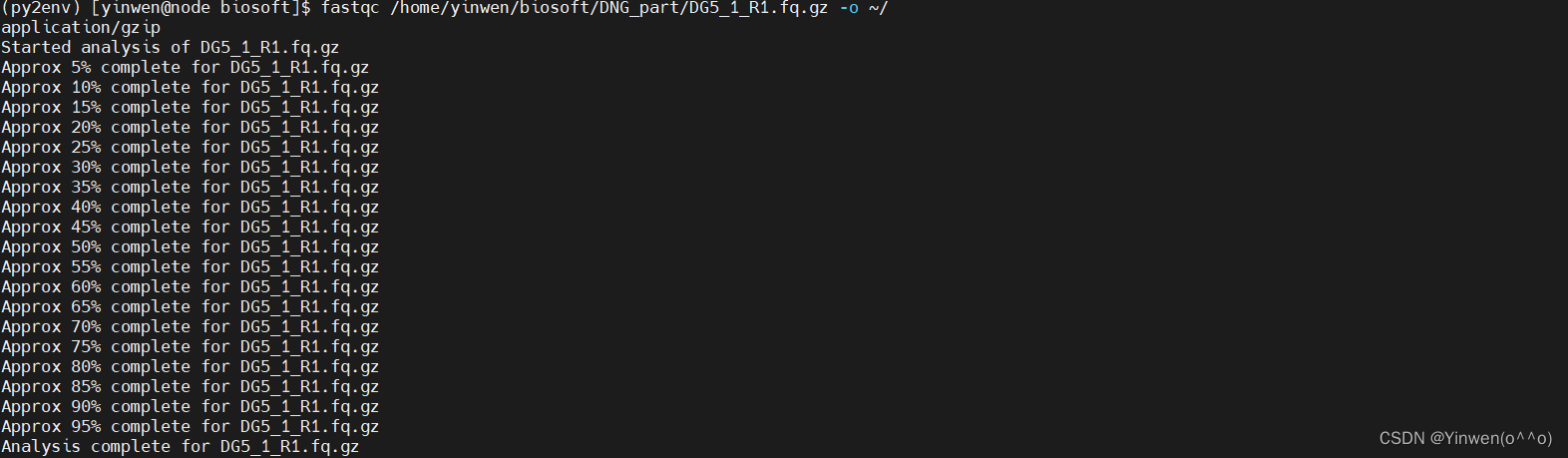

#然后批量跑
fastqc -t 15 /home/yinwen/biosoft/DNG_part/*fq.gz -o /home/yinwen/RNA-seq_report/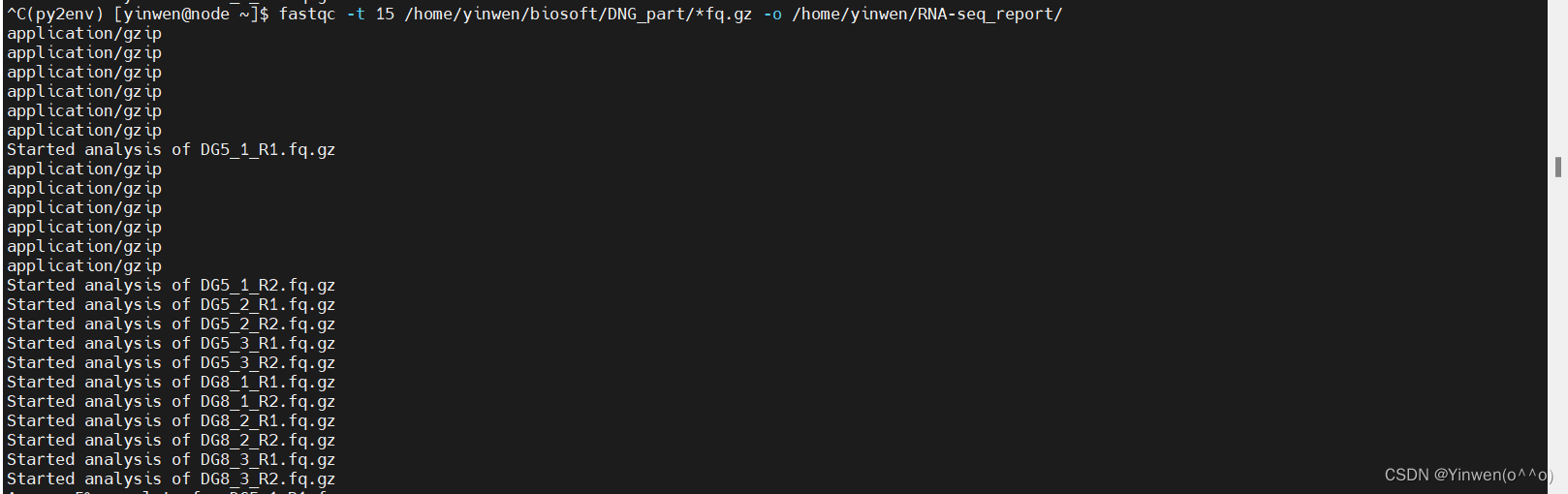
#打包报告
tar -cvf html_files.tar *.html














 275
275











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









