







在组装未知基因组时,往往需要利用重测序数据提前进行基因组调查,以获取其基因组规模,杂合率,重复序列比例,GC含量等信息。从而更好地拟定后继测序策略。基因组调查可以采用kmers方法。kmers基因组调查分为kmers频数统计和基因组评估两步。原理已经有大佬讲得很清楚啦:https://www.jianshu.com/p/94da86093843
这里以猕猴桃基因组hongyang为例,具体使用kmc+genomescope软件进行基因组调查。kmc会对重测序reads数据进行kmers库的构建,genomescope则根据kmers库进行基因组特征评估。我们来尝试复刻文章中的基因组调查结论。
1.下载基因组重测序数据
我们以猕猴桃hongyang为例,首先获取猕猴桃基因组重测序数据。进入NCBI网址即可查看具体信息:https://trace.ncbi.nlm.nih.gov/Traces/index.html?run=SRR9329821
找到对应的重测序数据编号SRR9329821。
服务器安装sra-tools后可以直接使用prefetch下载。
$ prefetch SRR9329821
2.sra文件转化为fastq文件
下载得到的数据约为11.3G,我们接下来使用fastq-dump将sra文件处理为fastq文件。
$ fastq-dump --gzip --split-3 SRR9329821/
这里对于不知道是单端测序还是上端测序的sra文件,可一律采用–split-3,程序会自动识别。这一步较为耗时,建议nohup挂载到后台运行。 运行结束后产生SRR9329821_1.fastq.gz和SRR9329821_2.fastq.gz两个双端文件。
3.fq文件质控
采用fastp软件进行文件质控。
此处-l代表过滤长度在36bp以下的reads,-w设定滑窗规模为6,以此标准计算平均reads质量,-q代表过滤平均质量Q20以下的reads,--compression表示压缩程度(1-9),越大代表压缩过程速度最慢,越小代表速度快。
$ fastp -i SRR9329821_1.fastq.gz -I SRR9329821_2.fastq.gz -o SRR9329821_1.fastq.cleandata.gz -O SRR9329821_2.fastq.cleandata.gz -l 36 -q 20 -n 6 -w 6 --compression=6
至此,重测序数据准备工作已经完毕,下面开始制备kmer数据库以及基因组调查。
需要用到软件KMC,smudgeplot和genomescope,请提前下载并安装。
三者都可以用conda下载:
$ conda install -c bioconda smudgeplot
$ conda install -c bioconda genomescope2
$ conda install -c bioconda kmc
安装完毕后可执行后继步骤。
4.构建重测序kmers库
在刚刚质控后的存有双端测序*fq.gz文件的文件夹下,利用双端测序数据构建kmer库。
$ mkdir tmp #建立临时文件夹
$ ls SRR9329821_1.fastq.cleandata.gz SRR9329821_2.fastq.cleandata.gz > KiWi
$ kmc -k21 -t32 -m64 -ci1 -cs10000 @KiWi test_kiwi tmp #运行kmc以构建kmer库,设定kmer构建长度为21,线程占用数为32,内存为64G。
$ kmc_dump -ci50 -cx3000 test_kiwi kmer21.dump #提取所有覆盖度在50X-3000X的kmers
kmc执行reads建库命令,运行完kmers建库后会生成kmers数据库,其数据分别保存于.kmc_pre和.kmc_suf两个文件。kmc_dump命令会产生.dump文件,里面存放有kmers的list。
5.匹配杂合kmers对
为了后继估计基因组杂合程度,需要提前统计杂合的kmers对。将上述结果.dump文件做为输入,运行后的结果保存于kmer_hongyang_coverages.tsv和kmer_hongyang_sequences.tsv两文件中。-o相当于指定两个tsv结果文件的标题。
$ smudgeplot.py hetkmers -o kmer_hongyang < kmer21.dump
至此为止所有数据准备工作已经结束啦,下面正式开始进行基因组特征调查。
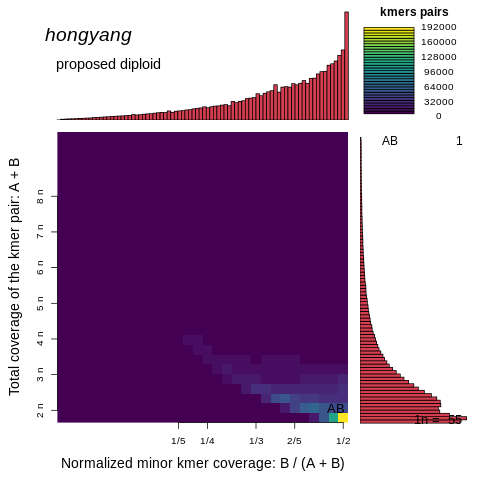
6.smudgeplot评估基因组倍型
smudgeplot软件可以以kmer_hongyang_coverages.tsv为输入可视化倍型,这里直接上代码。
$ smudgeplot.py plot -t "hongyang" -q 0.99 kmer_hongyang_coverages.tsv
7.genomescope评估基因组特征
Genomescope是2017年发表于bioinformatic的一个工具,以处理一些高复杂度的基因组调查。第一个版本仅能预测二倍体基因组,第二个版本可以预测多倍体基因组特征。Genomescope利用kmer频数统计结果,即KMC结果.hist文件进行基因组评估,过程如下:
$ kmc_tools transform test_kiwi histogram kmer21_hongyang.hist -cx10000
$ genomescope2 -i kmer21_hongyang.hist -k 21 -p 2 -o . -n hongyang_genomescope
得到的峰图可以查看kmers频数分布图,也可以得到summary.txt以查看基因组大小,杂合度,重复片段比例等详细信息。
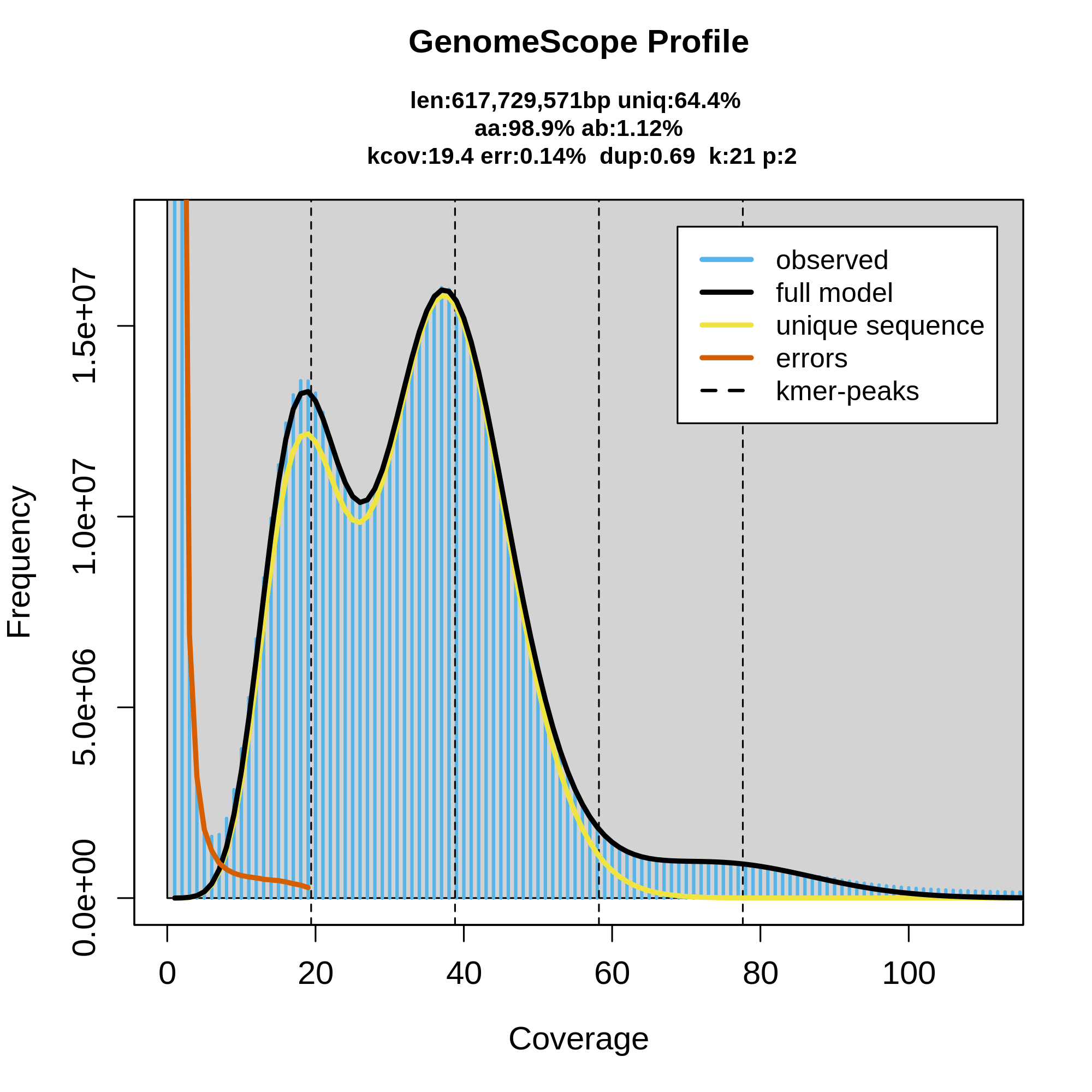
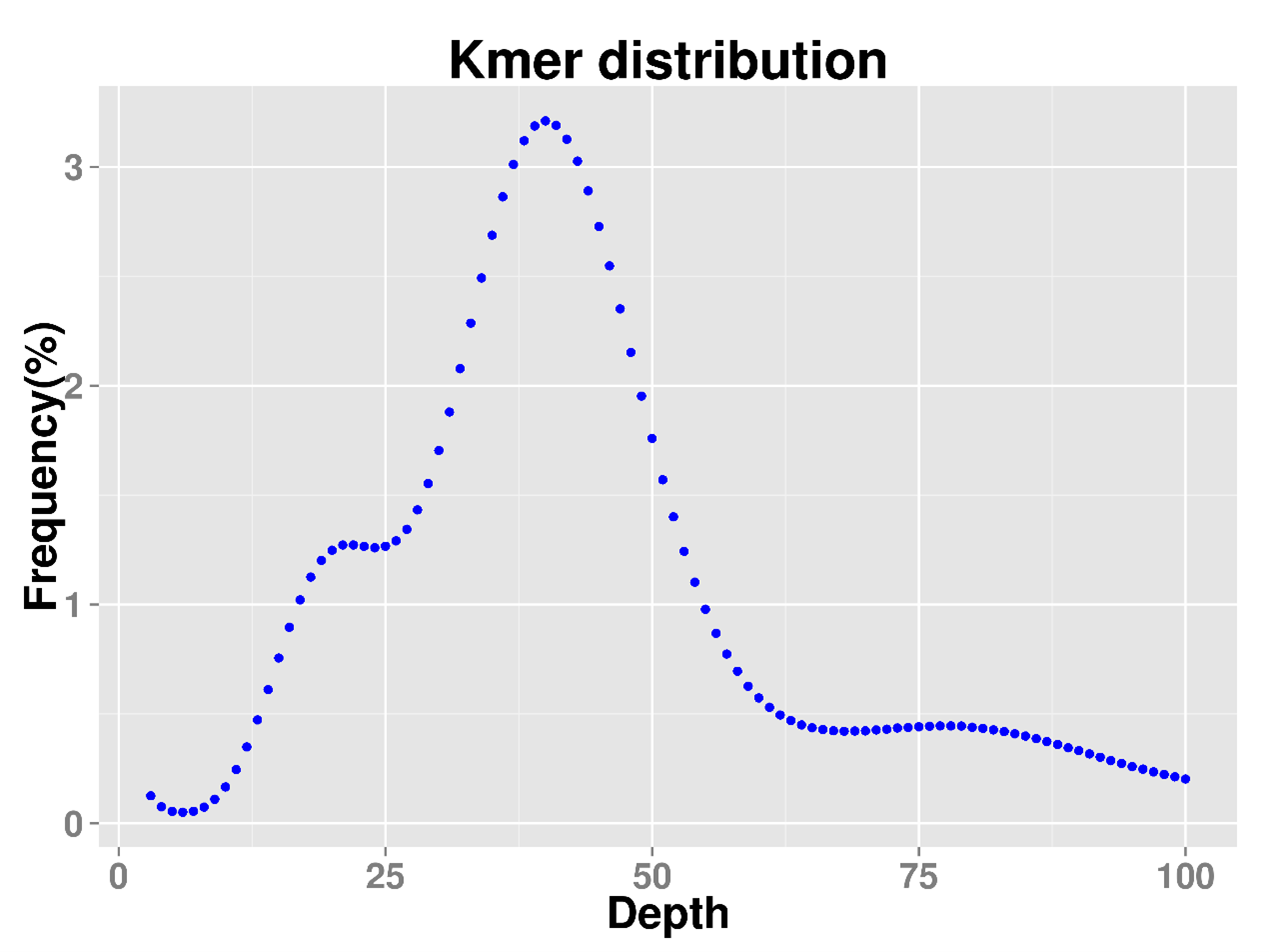
结果可见基因组预估大小为617M,杂合度为1.12%,重复率0.69。跟原文的结论还是非常接近的,只不过原文用的是19-mer进行的基因组调查,本文用的21-mer,可能造成一些结果出入。另外,genomescope作图的美观性还是不错的,推荐大家试一试。














 973
973











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









