







上周发了第一篇记录在知乎上面,主要是从raw data(fastaq)进行往下的分析处理,想着看文章有没有人看再考虑是否接着写。然后发现还是有不少人对这个生信分析感兴趣的,也有很多点赞关注的,也有问我有没有推荐教程啥的,这段时间我也大致翻了知乎和CSDN,常见的点赞比较多的文章,在我具体实操之后,发现它们写的遗失了很多分析细节,根本达不到科研工作的水平和要求,比如:
-
怎么正确读取GEO上格式不同的数据?(mtx,csv,tsv,txt等)
-
不同文件对应的数据是怎么存储在anndata对象中的?怎么直观的理解anndata数据格式?
-
scanpy的数据分析流程直观来说分为几个部分?对于一些方法,为什么要在这个地方用?(目的性说明缺失)
-
如何根据高可变基因的输出情况来反馈调整数据预处理?如何使用pyplot进行的可视化反馈修改?
-
如何在初步聚类分析后,对adata数据进行矩阵处理,切割出我需要的数据来进行下一步聚类分析?
这些问题我目前没有看到有写的很好的文章,也可能是写的很好的都要收费了?(这点开源精神都没有吗...)这周的学习是把之前大三上学的生信课的基础scanpy分析流程过了一遍,然后研究了一下数据库读取和数据预处理的细节问题,今天突然不想学习(开摆!),所以就来写一篇记录,希望能正儿八经的给到一定的学习帮助(如果觉得有用还请点个赞嘿嘿~)考虑到想学生信的人水平参差不齐,我就直接从最基础的环境开始写起(当然搭建环境其实可以参考别人的文章也是完全可以的)
上周的文章链接:生物信息学scRNA-seq和RNA-seq处理流程学习记录 - 知乎 (zhihu.com),至于我为啥写这些东西,原因也在这篇文章里,感兴趣也可以看一看。
(一)conda环境搭建
这部分内容我不打算细说,本来我使用服务器去写代码,但是发现scanpy其实在前期学习没必要去使用linux服务器,我自己的电脑(i78核16G,3060,6G显存)也完全够用,所以懒得折腾。如果是纯新手,请先下载vscode和anaconda,地址:Download Visual Studio Code - Mac, Linux, Windows,Download Now | Anaconda
然后就是安装和添加path环境变量(请务必记得添加环境变量,我个人认为通俗的解释就是,你不添加环境变量你后续就无法在其他目录下使用conda环境),然后在vscode上下载python,jupyter notebook的拓展,这个我就不写了,网上相关的文章很多,总之第一步你要先搞定能在vscode上书写.ipynb文件。
当然其实这一步对于纯生物的同学来说也不算简单,我当初就折腾了挺久....只能说take your time,不要着急慢慢来。搞定这些之后在conda prompt中创建一个新的虚拟环境,并安装这些包,然后在vscode创建一个scRNA-seq.ipynb的文件,选择你自己创建的虚拟环境作为内核,然后我们就可以开始第二步啦。
conda create -n czf python=3.11 # 将czf替换成你想使用的环境名称
conda activate czf
pip install scanpy
pip install jupyter notebook
pip install ipykernel(二)GEO数据下载和读取
我直接说我自己的工作流程,尽可能细致一点。现在D盘创建一个labwork_study的文件,然后再创建一个results文件,并在results文件中创建一个output.h5ad的文件用来存储后续分析的输出数据。然后我们前往GEO数据库,GEO Accession viewer (nih.gov),按照我下面这张图所示下载这四个文件:
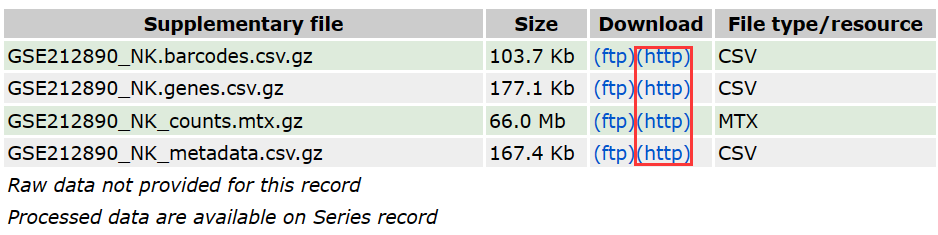
Fig1. 下载用于分析的原始数据
至于数据是否解压其实不影响,因为scanpy可以直接读取.gz文件,但是我自己解压了,所以为了方便你和我一样进行后续操作,你可以先自己创建一个transform.py的文件,然后使用下面的代码进行解压。你唯二需要做的事情是,确保你下载了os,gzip的包,并且修改正确的路径。
import os
import gzip
import shutil
# 指定源目录和目标目录路径
source_directory = r"D:/labwork_study"
target_directory = r"D:/labwork_study"
# 遍历源目录下的所有文件
for root, dirs, files in os.walk(source_directory):
for file in files:
if file.endswith(".gz"):
# 构建完整的源文件路径
gz_file_path = os.path.join(root, file)
# 构建解压后的文件路径(在目标目录下)
extracted_file_path = os.path.join(target_directory, file[:-3])
# 解压缩 .gz 文件并保存到目标目录
with gzip.open(gz_file_path, 'rb') as f_in:
with open(extracted_file_path, 'wb') as f_out:
shutil.copyfileobj(f_in, f_out)
print(f"解压完成:{gz_file_path} -> {extracted_file_path}")
最后创建在labwork_study目录下创建scRNA-seq.ipynb文件,并打开,先如下导入必需的库,并设置提醒层级和图像清晰度:
import scanpy as sc
import matplotlib.pyplot as plt
sc.settings.verbosity = 0
sc.logging.print_header()
sc.settings.set_figure_params(dpi=200, facecolor='white')指定产生的数据对应的存储文件的路径:
results_file = 'D:/labwork_study/results/output.h5ad'读取本次分析所需的四个文件并确保gene名唯一,这一步其实大有讲究,而且网上我没看到有啥很好的教程,如果有需要我可以单独再写一篇文章:
adata = sc.read_mtx(filename='GSE212890_NK_counts.mtx.gz', dtype='float32')
genes = sc.read_csv(filename='GSE212890_NK.genes.csv', dtype='str')
adata.var_names = genes.X[:,1] # 这个和它的读取有关,需要直接读取第二行的genes名称
adata.obs_names = sc.read_csv(filename='GSE212890_NK.barcodes.csv', dtype='str').obs_names
adata.obs = sc.read_csv('GSE212890_NK_metadata.csv',dtype='str').obs
adata.var_names_make_unique()
adata(三)数据预处理(质量控制)
第一步质控,先处理掉那些明显偏差过高的细胞
# 第一步质控:删除明显错误的数据
sc.pp.filter_cells(adata, min_genes=200) # 过滤掉那些表达基因数少于200的细胞
sc.pp.filter_genes(adata, min_cells=3) # 过滤掉在少于3个细胞中表达的基因
adata # 检查删除后的adata数据量第二步,对于那些严重影响后续高可变基因筛选的管家基因单独拿出来,分析其占比和协方差影响,并在最后消除其影响:
# 确定基因功能请前往ensembl和NCBI
adata.var['mt'] = adata.var_names.str.startswith('MT-')
adata.var['rp'] = adata.var_names.str.startswith('RP')
adata.var['ctb'] = adata.var_names.str.startswith('CTB')
adata.var['mt']
adata.var['rp']
adata.var['ctb']
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
sc.pp.calculate_qc_metrics(adata, qc_vars=['rp'], percent_top=None, log1p=False, inplace=True)
sc.pp.calculate_qc_metrics(adata, qc_vars=['ctb'], percent_top=None, log1p=False, inplace=True)
adata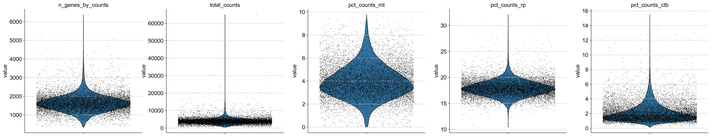
Fig2. 小提琴图来观察数据分布
绘制散点图,同样可以直观的看出组分分布,并协助判断哪些需要删除
fig, axes = plt.subplots(1, 4, figsize=(20, 5)) # 1行4列,整体图形大小为20x5英寸
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts', ax=axes[0], show=False)
axes[0].set_title('Total Counts vs N Genes by Counts')
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt', ax=axes[1], show=False)
axes[1].set_title('Total Counts vs Pct Counts MT')
sc.pl.scatter(adata, x='total_counts', y='pct_counts_rp', ax=axes[2], show=False)
axes[2].set_title('Total Counts vs Pct Counts RP')
sc.pl.scatter(adata, x='total_counts', y='pct_counts_ctb', ax=axes[3], show=False)
axes[3].set_title('Total Counts vs Pct Counts CTB')
plt.tight_layout() # 调整子图间距
plt.show()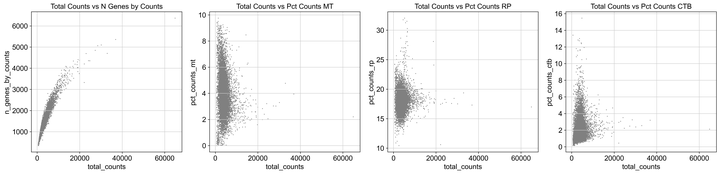
Fig3. 散点图辅助判断筛选
根据散点图情况,进行第二次筛除
adata = adata[adata.obs.n_genes_by_counts < 3800, :] # 具体数值需要根据散点图进行判断
adata = adata[adata.obs.pct_counts_mt < 5, :] # 分别删除基因表达过高和Mt表达占比过高的
adata = adata[adata.obs.pct_counts_rp < 20, :]
adata = adata[adata.obs.pct_counts_ctb < 6, :]
adata接着绘制读长情况,来辅助判断后续归一化中target_sum,即我们进行归一化的目标数量进行参考,一般选取中位数作为target_sum。
sc.pp.calculate_qc_metrics(adata, inplace=True)
plt.figure(figsize=(8, 6), dpi=500)
plt.hist(adata.obs['total_counts'], bins=50)
plt.title('Total Read Counts Distribution')
plt.xlabel('Total Counts per Cell')
plt.ylabel('Frequency')
plt.show()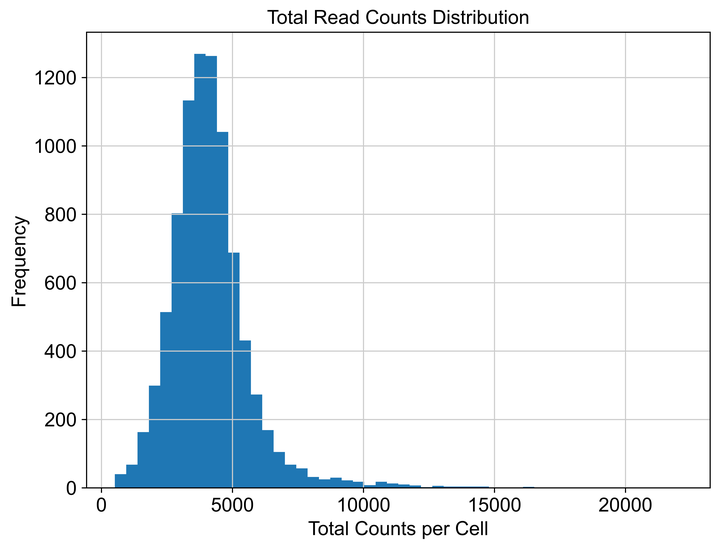
Fig4. 为归一化参数辅助判断
接着进行归一化,抹平整体的表达差异;使用对数,来缩小基因表达差异上过于离谱的数值大小;同时,我们设置高可变基因的定义,根据最大最小值和离散度来进行确定,这个设置数值可以参考我的。
# 根据上图对每个细胞的总表达量进行归一化校正
sc.pp.normalize_total(adata, target_sum=4800)
# 取对数来减小夸张的差异性,便于后续计算
sc.pp.log1p(adata)
# 基于最大最小值和离散度设置高变基因的筛选标准,这个数值设置我也是参考了文献的
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)针对上述操作的结果,使用散点图来可视化展示
sc.pl.highly_variable_genes(adata) # 可视化查看结果
# 在进一步筛选之前存储原始数据
adata
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
adata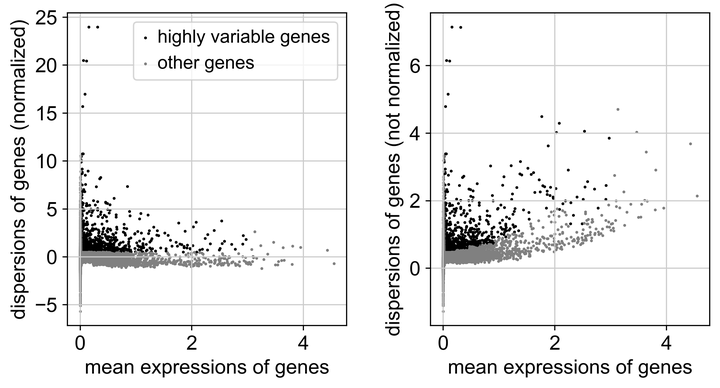
Fig5. 观察高可变基因占比情况
第三步,进行主成分分析,先进行校正,再观察各个PC的占比,来为下一大步骤的主成分数量选取做一个参考:
sc.pp.pca(adata, svd_solver='arpack')
sc.pl.pca_scatter(adata, color='total_counts', title='Before Regress Out')
# 进行校正
sc.pp.pca(adata, svd_solver='arpack')
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt','pct_counts_rp','pct_counts_ctb'])
# 校正后的PCA
sc.pp.pca(adata, svd_solver='arpack')
sc.pl.pca_scatter(adata, color='total_counts', title='After Regress Out')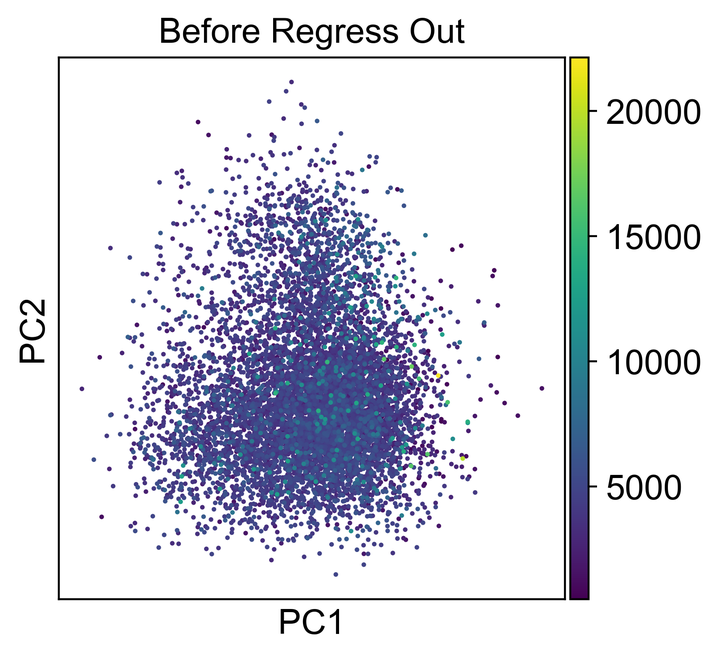
Fig6. 观察降维后数据分布情况
# 观察PC占比,为后续计算细胞的neighborhood relations时应考虑多少个PC提供参考
sc.pl.pca_variance_ratio(adata, log=True)
# 运行PCA
sc.pp.pca(adata, svd_solver='arpack')
# 计算累积方差解释
cumulative_var_ratio = adata.uns['pca']['variance_ratio'].cumsum()
# 设定方差解释的阈值
threshold = 0.9 # 例如,设定阈值为90%
num_components = (cumulative_var_ratio >= threshold).argmax() + 1
# 输出结果
print(f"Number of principal components to reach {threshold*100}% variance: {num_components}")
print("Cumulative variance explained by each component: ", cumulative_var_ratio)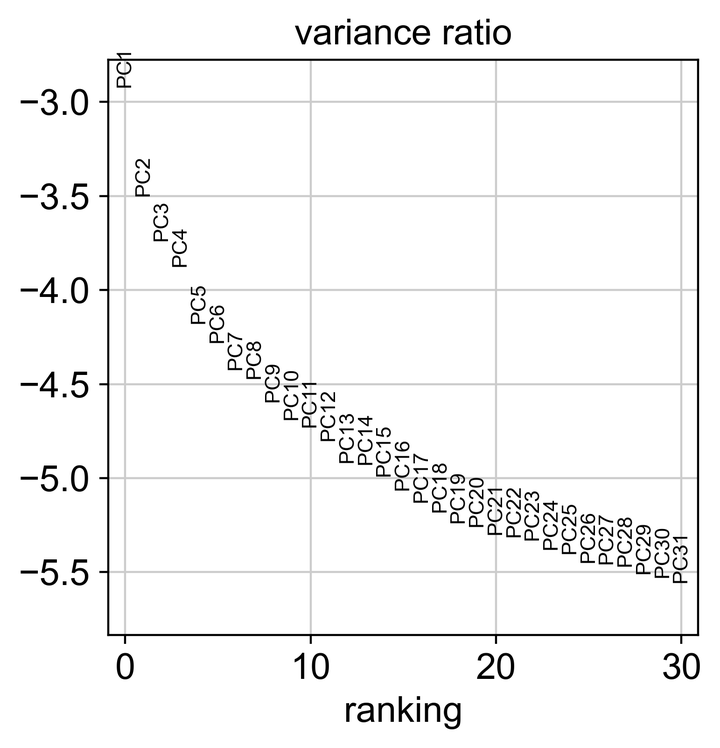
Fig7. 观察PC成分对于解释的效果占比
完成基础的数据预处理,保存数据。
adata.write(results_file)
adata(四)降维聚类和根据差异基因确定细胞类群
首先使用neighbors方法计算单细胞数据中的近邻关系,这是后续分析步骤(如聚类、降维等)的基础
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40) # 用数据矩阵的PCA表示来计算细胞的邻域图
adata使用umap降维方法,来观察前期数据预处理之后的效果,同时可以观察一下自己比较关心的基因的表达情况(是否在某一块区域集中发生了表达,说明预处理做的很好)
sc.tl.umap(adata)
sc.pl.umap(adata, color=['NCAM1','NKG7','FCGR3A',"KLRF1"])
sc.pl.umap(adata, color=['FCGR3A',"KLRF1"], use_raw=False)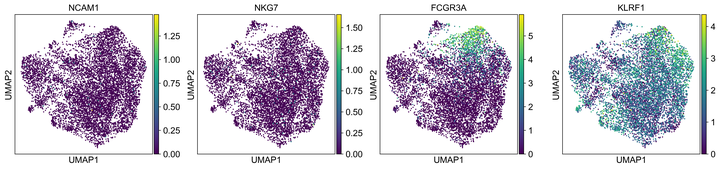
Fig8. 观察你最关心的几个基因的表达分布情况
接着我们使用leiden算法,进行细胞聚类分析,下面这段代码需要你自己根据自己实际情况来调整resolution大小,我这里做的是初步聚类,所以0.3就差不多可以了,如果你是想做很细致的聚类,就可以调大这个数值。这个数值直观上简单的理解,就是越大分的越细致,越小分的越粗糙;在初步聚类的时候我们就不需要那么大,一般0.3~0.5是比较好的。然后使用umap降维绘制结果。
sc.tl.leiden(adata,resolution=0.3)
sc.pl.umap(adata, color=['leiden', 'ISG15', 'RP11-54O7.11', 'TNFRSF4'])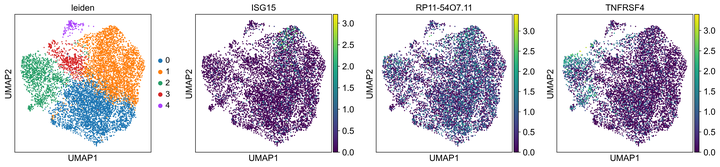
Fig9. 初步聚类的结果,同时观察感兴趣的基因表达情况
接着将结果写入预先准备好的.h5ad文件。
adata.write(results_file)接着就是查找差异基因,直接用我的代码就完事了
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# 确定要提取的基因数量
n_genes = 25
# 获取差异表达基因的结果
groups = adata.uns['rank_genes_groups']
group_names = groups['names'].dtype.names
# 遍历每个群组并提取前 25 个基因
for group in group_names:
gene_names = groups['names'][group][:n_genes]
print(f"群组 {group} 的前 {n_genes} 个差异表达基因:")
print(gene_names)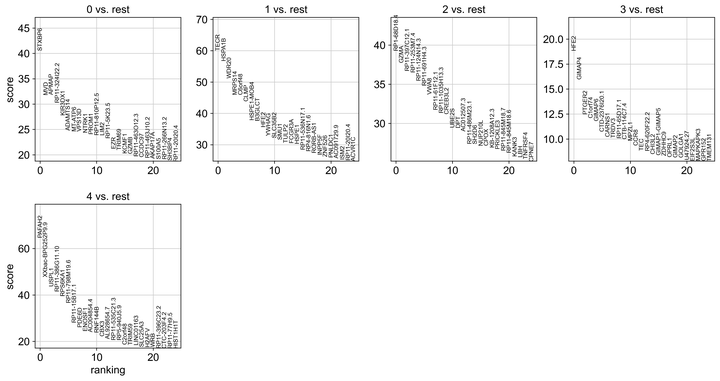
Fig10. 输出每个类群的主要差异基因并展现表示差异程度
然后你就需要动用你的生物学知识了,根据前25名的差异基因的情况,来判断每个聚类群体到底对应什么细胞,比如说是NK细胞,还是B细胞,或是T细胞等等。我的这个代码的结果后面还需要重新调整,如果你生物知识很猛,一眼就能看出来哪些基因明显是对应哪一种细胞类群,那这一步就完成了,下面手动写入细胞注释,因为我这里后续还要修改,所以下面这个只是示范,告诉你该怎么写:
new_cluster_names = [
'1: CD14+Monocytes',
'2: NK',
'3: Megakaryocytes'
]
adata.rename_categories('leiden', new_cluster_names)最后用tracksplot验证基因与细胞的关系
sc.pl.rank_genes_groups_tracksplot(adata, n_genes=3)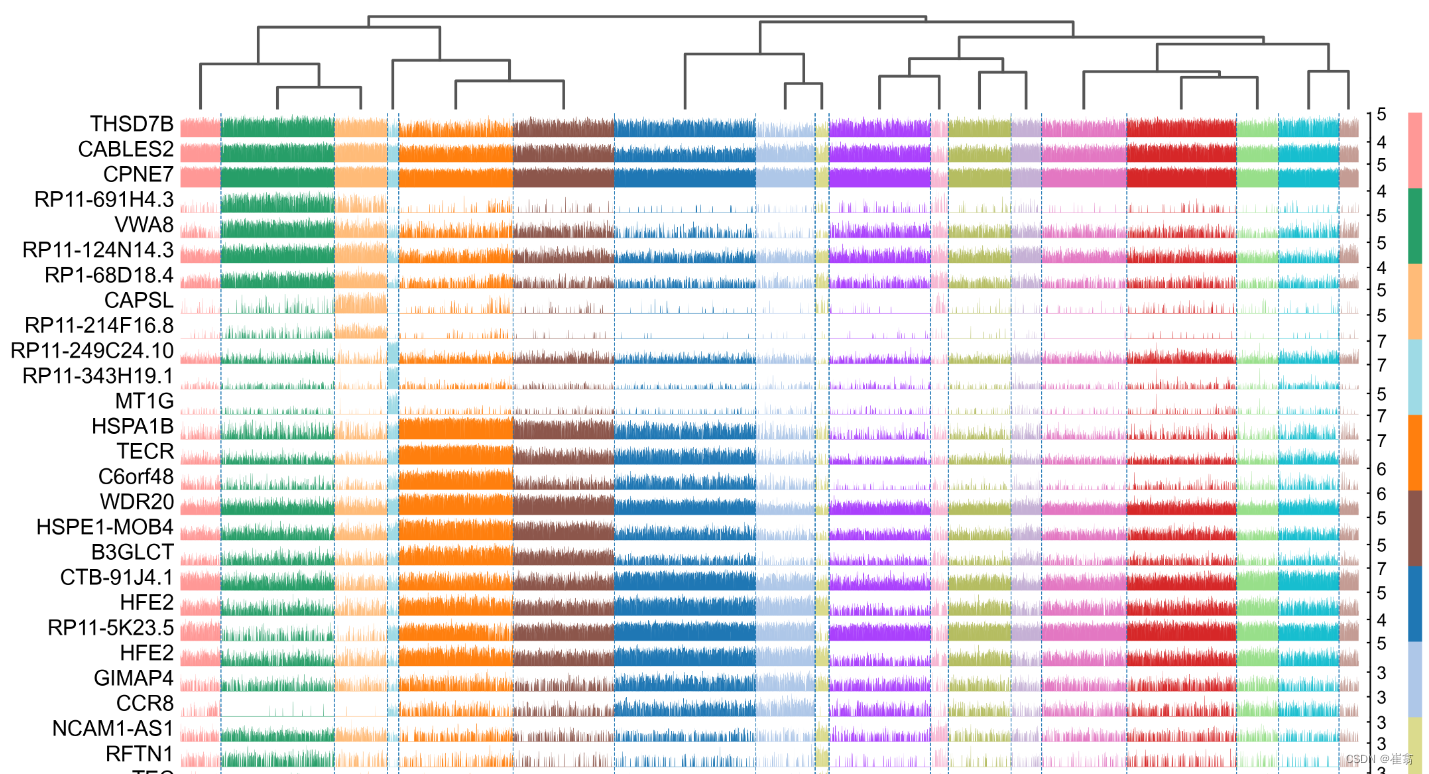
(五)总结和下一次工作内容的展望
如果你完成了上述流程,恭喜你,你已经完成了最基础的一个scanpy的下游分析流程了,接下来你就可以深究以下问题:
-
尽管我在预处理阶段已经对MT,RB基因进行了筛除,并消除了协方差效果,但还是在最后的差异基因中出现很多干扰,这说明我的代码很可能在前面的参数设置或是思路上存在一定的问题;
-
你可以进一步探索初筛应该在什么样一个程度。这一点你可以通过每一次处理之后的adata数据矩阵大小来观察在你的质量控制下,有多少细胞和基因被你噶掉了。通过反复的调试,你也一定能找到一个比较合理的范围;
-
进阶操作:首先你需要去阅读scanpy自己的官方手册,这将受益匪浅(我就是看手册写的代码);其次,去熟悉一下matplotlib的个性化绘图会对未来科研作图有不小的帮助;以及学习numpy和pandas处理数据尤其是矩阵的操作方法,这将在后续的进阶操作中起到重要的作用。
-
你还需要熟悉GEO数据库上各种数据类型,了解每种文件的读取方式;同时你要能够在NCBI和Ensembl中查找你感兴趣的基因,并判断它对应的细胞类群,同时你也需要参考相关文献来辅助判断;
总结一下,scRNA-seq的分析流程绝不是简单的调包,而是在调包的过程中,你通过不断的调试代码,不断的绘制新的可视化图像,来不断深化你对scRNA-seq的理解。我对生信分析的态度是,它已经是科研工作必不可分的一部分,只有既做好湿实验,也做好干实验数据分析和可视化,才算是符合时代发展潮流的生物领域的学者。
以及我的代码依旧是在很多参数或是逻辑上可能存在错误甚至谬误,如果你发现不合理的地方,请一定告诉我,我也会非常感谢!如果你对这个流程依旧有问题,欢迎评论区留言。
最后,如果觉得我写的还不错就点个赞吧~















 1059
1059

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









