






点击蓝字 关注我们
从基因组功能学角度揭示肠菌对复杂疾病的潜在影响

https://doi.org/10.1002/imt2.14

3.29
iMeta
REVIEW ARTICLE
● 2022年3月29日,南京医科大学陈连民等在iMeta在线发表题为“Decoding microbial genomes to understand their functional roles in human complex diseases”的综述文章。
● 该综述系统总结了微生物在人类复杂疾病中的潜在功能机制并介绍了用于解析微生物基因组的常用工具。同时也介绍了领域内用以验证微生物潜在功能的前沿技术,帮助我们更好理解微生物在人类复杂疾病中的作用,推动基于肠菌的精准医疗发展
● 第一作者:王屹丰(南京医科大学第一附属医院)
● 通讯作者:陈连民(南京医科大学第一附属医院/格罗宁根大学医学中心)(lianminchen@njmu.edu.cn)
● 通讯作者:孔祥清(南京医科大学第一附属医院)
(kongxq@njmu.edu.cn)
● 合作作者:董权斌, 胡世贤, 邹花一阳, 吴婷婷, 史倞, 张海锋, 盛燕辉, 孙伟
摘 要
心血管病、肥胖、炎症性肠病、肾病、2型糖尿病和癌症等复杂疾病已经成为公共卫生健康的主要负担,影响全球超过20%的人口。复杂疾病的病因仍未被完全研究清楚,但是,传统上认为是基因和环境(比如:饮食习惯)共同作用的结果。此外,近些年的研究表明肠道微生物可能通过其代谢分子影响人类健康。因此,解码微生物基因组对揭示其潜在的功能非常重要。在这篇综述中,我们系统总结了微生物在人类复杂疾病中的潜在功能机制并介绍了用于解析微生物基因组的常用工具。此外,还介绍了领域内用以验证微生物潜在功能的前沿技术,以帮助我们更好理解微生物在人类复杂疾病中的作用,从而推动基于肠菌的精准医疗发展。
关键词:肠道微生物组,代谢物,复杂疾病,宿主-环境互作
亮 点
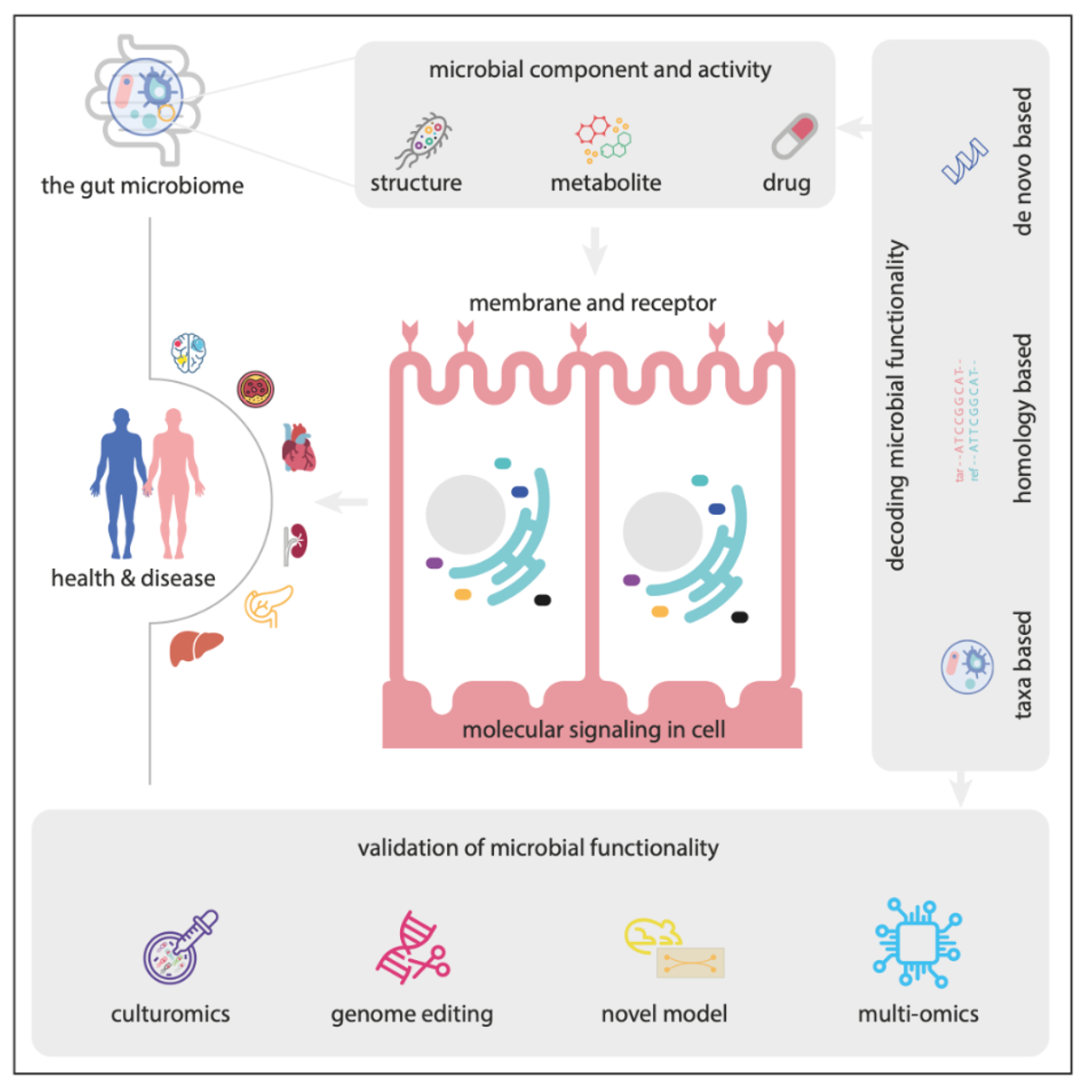
● 肠道微生物组在人类健康和疾病中的功能角色
● 解码微生物组功能的通用工具
● 验证微生物组功能的前沿技术
视频解读
Bilibili:https://www.bilibili.com/video/BV1HT4y1r7pq/
Youtube:https://youtu.be/UwV1nvo2aNw
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
全基因组关联分析(GWAS)已经解析了人类复杂疾病的遗传机制,极大地促进了我们对疾病病因的理解,同时也促进了基因疗法的发展。然而,个体罹患复杂疾病病因复杂,遗传学仅占其有限的一部分。例如,GWAS表明遗传因素分别只占患2型糖尿病和克罗因病的6%和20%。近年来,肠道微生物组在人类复杂疾病发展中的作用逐渐得到认可,并且成为一个蓬勃发展的研究领域。
人类肠道定植了大量的细菌、古菌、真核生物以及病毒,这些微生物总数和人体内细胞数量相当,统称为“微生物组”。肠道微生物组参与消化食物、训练宿主免疫系统、调节肠道内分泌功能、神经信号转导、改变药物代谢以及清除毒素等反应,此外还会产生大量影响宿主的化合物。在以小鼠为对象的研究中,肠道微生物在无菌动物模型发展至炎症性肠病(IBD)中至关重要。在人类研究中,患有免疫系统相关疾病母亲的婴儿呈现出不一样的肠道微生物组成,可能进一步引起适应性免疫反应。所有的证据都表明了肠道微生物组在复杂疾病发展中的关键作用。
宏基因组学测序技术的快速发展和大型队列研究使我们能够将肠道微生物组与宿主临床表型相结合,从而识别候选疾病相关的微生物特征。近年来,大量的肠道微生物组成和复杂疾病之间的相关性研究陆续发表,包括但不限于心血管疾病、糖尿病、炎症性肠病、过敏以及癌症。与人类基因组不同,肠道微生物组有一定的可操作性,因此肠道微生物组正在逐渐成为疾病预防和治疗的靶点。然而,学术研究与临床转化之间仍存在巨大距离,比如不同研究之间的疾病特异性微生物组缺乏一致性,因果推断较差,以及当前基于微生物组的治疗在患者中的效率不尽人意(例如,粪便微生物组移植(FMT)和益生菌的使用)等。这可能是由于1)许多肠道细菌具有不确定性,甚至在特定条件下会表现出相反的表型;2)大部分研究都聚焦于微生物组成,但这是远远不够的,因为不同的亚型表型也不尽相同。基于解码微生物基因组的功能分析发现,cutC/D等微生物基因参与苯乙酰谷氨酰胺和氧化三甲胺(TMAO)的合成,而这两种代谢物是诱发CVD的危险因素。因此,除了肠道微生物组成,我们更应该深入进行功能分析,从而解决以上问题。
本文综述了近年来与人类健康和疾病相关的肠道微生物组研究进展,特别关注其潜在功能,主要包括微生物膜的毒力因子、衍生的小分子以及药物代谢。与此同时,常用解码微生物基因组功能的分析工具也被提及。最后,我们还介绍了领域内的前沿技术,或许可以帮助我们更好理解微生物组在人类复杂疾病中的机制,促进基于肠道微生物组的个性化医学发展。
影响人类健康和疾病的微生物功能
特殊的菌体结构
肠道微生物往往具备耐药性和毒性的基本结构,比如鞭毛、菌毛、荚膜、孢子及生物膜等,是肠道内数以亿计细菌生存和活动的基础,其对宿主的直接影响可以归因于此(图1)。
菌毛是从细菌细胞壁产生的直丝。鞭毛比菌毛长,可以旋转并产生推力,推进细菌向前移动。菌毛和鞭毛的形成往往由基因簇编码,且不同物种之间差异很大。例如,在菲姆沙门氏菌和大肠杆菌中分别鉴定出10个和6个鞭毛形成基因。即使两者都可以侵染宿主,但背后的机制不尽相同。许多细菌病原体都是靠机动性来感染宿主,因此鞭毛在这一过程中起着关键作用。与鞭毛不同,菌毛携带毒力因子,帮助细菌粘附在人类细胞上。例如,百日咳博德氏菌利用其黏附素与纤毛呼吸细胞结合,引起百日咳。淋病奈瑟菌的菌毛帮助其与宫颈细胞和颊部细胞结合,引起淋病。如果没有菌毛帮助结合人肠道上皮细胞,大肠杆菌和空肠弯曲杆菌就不能引起腹泻。
荚膜是位于细胞膜外的多糖层,也是细菌细胞外膜的一部分。荚膜普遍存在于革兰氏阴性菌和革兰氏阳性菌中,和仅在革兰氏阴性菌中发现的脂多糖和脂蛋白形成的二层脂膜不同。荚膜可以保护细菌免受机械损伤和环境变化(如温度、干燥、噬菌体和真核细胞)的危害。它还有助于细菌粘附在光滑的表面上。例如,可以导致龋齿的变形链球菌通过其荚膜附着在牙齿表面。荚膜也是病原微生物入侵宿主免疫系统并防止它们被巨噬细胞和中性粒细胞吞噬的必要条件。一项研究表明,肺炎链球菌荚膜的厚度与脑膜炎的严重程度相关。在真菌感染期间,荚膜与真菌细胞壁上的β-葡聚糖相互作用,激活宿主dectin-1相关的CARD9信号通路,从而诱导炎症。然而,荚膜材料也被成功应用于疫苗接种,如肺炎链球菌疫苗和流感嗜血杆菌疫苗。
不同于荚膜,孢子是一类异常坚固的细胞并能保护细菌免受极端条件的影响。因此,孢子可以保护致病菌免受抗生素等影响,从而产生毒力因子。芽孢杆菌和梭状芽孢杆菌是最常见的产生孢子的细菌,并可诱发多种感染性疾病。例如,蜡样芽孢杆菌的孢子可以在不同温度下存活从而引起食源性疾病。炭疽杆菌的孢子通过产生炭疽毒素和形成多聚γ-d-谷氨酸,引起皮肤和胃肠道吸入炭疽,从而保护细菌逃避免疫监视避免被吞噬。
生物膜可以被认为是一类能够自产胞外聚合物基质的细菌菌落,能够保护细菌细胞免受外部不利因素的影响,如温度变化、脱水和杀菌剂等。细菌生物膜通常具有致病性,据估计,高达80%的人类微生物感染,包括心内膜炎,囊性纤维化,牙周炎,鼻窦炎,骨髓炎,未愈合的慢性伤口,脑膜炎,肾脏感染和植入式装置相关感染等,都与生物膜形成相关。许多细菌都可以形成生物膜,最常见的包括大肠杆菌、粪肠球菌、金黄色葡萄球菌、表皮葡萄球菌、绿色链球菌、肺炎克雷伯菌、奇异变形杆菌和铜绿假单胞菌等。值得注意的是,金黄色葡萄球菌和表皮葡萄球菌会导致约50%的假体心脏瓣膜感染、70%的导管生物膜感染和80%的血流感染。不幸的是,单独使用抗生素治疗对生物膜引起的相关感染没有效果。这是因为1)生物膜可以延缓甚至阻止抗生素的渗透2)细菌可以通过水平基因转移获得抗性3)使用药物外排泵将抗生素从成熟的生物膜中泵入细胞外基质中。此外,生物膜还可以通过分泌c-di-NMPs来激活先天免疫系统诱发免疫反应,随后激活1型IFNs。
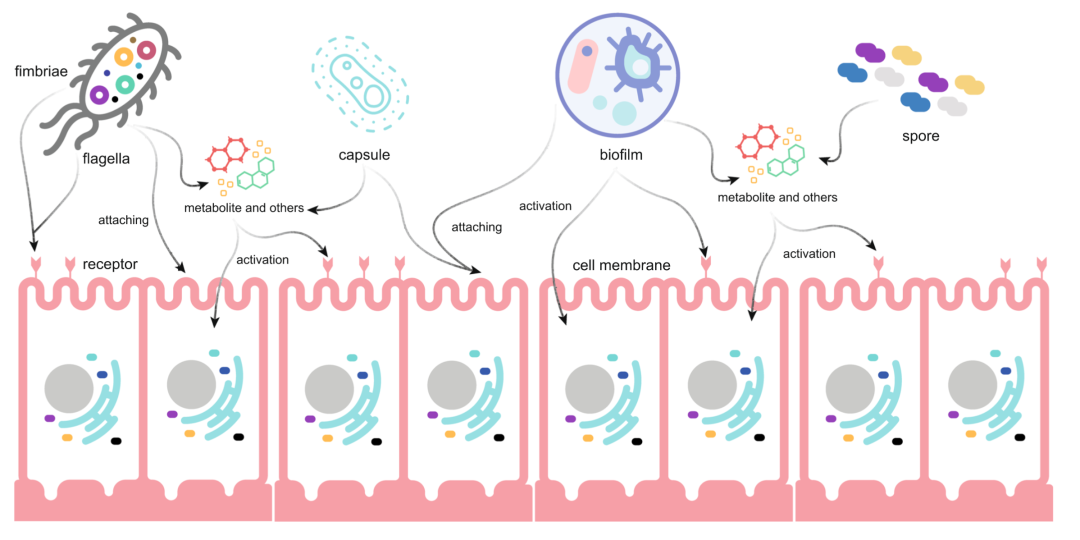
图1:引起微生物耐药性和毒性的基本结构
肠道微生物往往具备耐药性和毒性的基本结构,比如鞭毛、菌毛、荚膜、孢子及生物膜等结构,是肠道内数以亿计细菌生存和活动的基础,直接对宿主造成影响。特定的微生物结构可以帮助其附着人类细胞或受体,从而进一步激活各种信号通路。此外,它们还可能还具有抗生素耐药性、毒力因子、各种代谢物以及其他疾病相关分子
代谢分子
肠道微生物参与了一系列具有生物活性的代谢物合成和转化,这些代谢物可以作为底物和信号分子,参与人类生理功能或者诱发复杂疾病。微生物相关代谢分子主要包括短链脂肪酸(SCFAs)、氨基酸(AAs)、维生素、胆汁酸(BAs)、毒素、花青素以及植物学雌激素等(图2)。
短链脂肪酸(SCFAs)可以由结肠中的微生物通过发酵碳水化合物(如葡萄糖、淀粉和纤维等)或AAs(如赖氨酸、精氨酸、甘氨酸、亮氨酸、缬氨酸和异亮氨酸)进行合成。肠道微生物衍生的短链脂肪酸主要包括乙酸、丙酸、丁酸、戊酸和己酸。值得注意的是,乙酸、丙酸和丁酸占总SCFAs的95%以上,且在人体肠道中的摩尔比约为60:20:20。乙酸的生物合成主要依赖于编码磷酸转乙酰酶或乙酸激酶的基因。对于丙酸,则主要是由参与琥珀酸代谢途径、丙烯酸酯代谢途径和丙二醇代谢途径的基因编码。丁酸的生物合成主要来源于丙酮酸代谢途径、4-氨基丁酸代谢途径、戊二酸代谢途径以及赖氨酸代谢途径。丁酸的生产者包含很多物种,以厚壁菌门为主,包括粗粪杆菌、真杆菌、粪球菌以及罗斯伯里亚属,由丁酸激酶或乙酸辅酶A转移酶介导。双歧杆菌的基因组含有碳水化物物水解酶,参与乙酸和乳酸的生物合成。然而,也有实验表明双歧杆菌生产SCFAs的能力高度依赖基因含量的多样性。粘性阿克曼氏菌是一种可以诱导抗炎细胞因子分泌和增强肠道黏膜屏障的有益细菌,该菌可以通过粘蛋白降解产生SCFAs。
短链脂肪酸控制着人体肠腔内的PH,是一种重要的原料,与人类健康和疾病直接相关。肠道产生丁酸可以改善口服葡糖糖耐量试验后的胰岛素反应,而丙酸产生或吸收异常会增加患T2D的风险。SCFAs在人类生理过程中发挥重要作用的机制可能是它们在激活游离脂肪酸受体(FFARs、FFAR2和FFAR3)、G蛋白偶联受体(GPCRs、GPR109A和GPR42)、嗅觉受体(ORs、OR51E1和OR51E2)、过氧化物酶体增殖物激活受体-γ(PPARγ)和芳基烃受体(AhR)方面的信号传递能力。这些受体被乙酸、丙酸和丁酸激活后,可进一步导致信号级联反应,包括磷脂酶C(PLC)、丝裂原活化蛋白激酶(MAPK)、磷脂酶A2(PLA2)和核因子-κB(NF-κB)通路。这些通路在饱腹感调节、能量获取、脂肪存储、脂肪炎症以及神经系统发挥重要功能,因此SCFAs可以引起各种复杂疾病。此外,细胞内的SCFAs还可以影响组蛋白(主要是3和4)的乙酰化和去乙酰化(主要是赖氨酸残基N端的ε氨基酸上),从而影响基因的转录。这一过程主要是通过抑制组蛋白去乙酰化酶(HDACs)的活性导致更多具有转录活性的染色质,或增加组蛋白乙酰转移酶(HATs)的活性,从而激活乙酰化反应。HDACs和一系列复杂疾病都相关,包括结肠直肠癌和阿尔茨海默病。丁酸、丙酸和乙酸对HDACs都有抑制作用,其中丁酸的抑制作用最强。因此,肠道内的短链脂肪酸是引起人类复杂疾病的重要机制之一。
氨基酸(AAs)可以由肠道微生物通过消化食物蛋白质产生或通过从头合成。重要的是,所有9种必需氨基酸(组氨酸、赖氨酸、蛋氨酸、苯丙氨酸、苏氨酸、色氨酸、异亮氨酸、亮氨酸和缬氨酸)都可以由肠道微生物通过草酰乙酸/天冬氨酸合成基因合成。研究表明,操纵微生物基因组,比如产孢梭菌的fldC可以改变人类血液中芳香族氨基酸的含量。除了作为蛋白质合成和SCFAs发酵的底物外,AAs的缺乏也与人类疾病相关。其中,色氨酸是化学性质最复杂的氨基酸,对与宿主和微生物的代谢途径都有影响。色氨酸脱羧酶可以诱导色氨酸转化为色氨胺及其衍生物,许多细菌基因组中都含有该基因,包括乳酸菌属、消化链球菌属、拟杆菌属及双歧杆菌属。下游的代谢物可以被宿主不同的肠道受体感知,从而参与调节不同的代谢途径。这些受体包括GPR35、AhR、5-羟色胺受体(5-HT4R和5-HT3R、过氧化物酶体增殖物激活受体-γ共激活剂1α(PGC-1α)和孕烷X受体(PXR),与大脑、骨骼肌、胰腺和肾脏疾病相关。组氨酸可以通过激活p38γ-p62-mTORC1通路来影响T2D中的胰岛素信号传导。苯丙氨酸与帕金森等神经系统疾病相关,可以从多巴胺衍生。赖氨酸、蛋氨酸和苏氨酸则来源于草酰乙酸/天冬氨酸生物合成途径,该途径可以通过线粒体sirtuin4(SIRT4)、成纤维细胞生长因子21(FGF21)和丝氨酸/苏氨酸蛋白激酶25(STK25)参与胰岛素分泌和葡萄糖代谢。此外,亮氨酸、异亮氨酸和缬氨酸是与胰岛素抵抗和葡萄糖耐受不良相关的支链氨基酸(BCAAs),然而,其背后的机制尚不清楚。
毒素类可以由肠道微生物从多种底物合成,包括氨基酸和胆碱类化合物。可以和蛋白质结合的尿毒素如TMAO、吲哚、对甲酚、苯酚及其硫酸盐、葡萄糖醛酸酯、多胺以及马尿酸都是由肠道微生物从氨基酸中衍生的。TMAO及其衍生物主要由编码胆碱TMA裂解酶(cutC/D)、肉碱单加氧酶、甜菜碱还原酶和TMAO还原酶的微生物基因合成。氨基酸衍生出的尿毒症毒素如吲哚、对甲酚和苯酚的产生在很大程度上取决于不同菌群的基因含量。例如,编码色氨酸酶的基因在拟杆菌属中不尽相同,因此,只有部分拟杆菌属的菌种可以产生硫酸吲哚酚。这些毒素可以通过NF-κB、MAPK和JunN-端激酶途径进一步诱导慢性肾病(CKD)和CVD,从而启动促炎细胞因子和粘附分子的转录,导致炎症和氧化应激反应。从胆碱类化合物中衍生的毒素主要包括TMAO及其衍生物,其在CVD中具有多种作用,可以通过MAPK和NF-κB信号以及NLRP3炎症小体发挥作用,导致炎症。此外,革兰氏阴性菌(主要是拟杆菌)可以合成毒素脂多糖(LPS),通过NF-κB途径影响CAD的发生。
维生素是人类必须的营养素,且只能从食物和肠道微生物等外源性物质中获取。肠道微生物主要合成维生素K和大部分水溶性B族维生素,如如生物素(H)、钴胺素(B12)、叶酸(B9)、烟酸(B3)、泛酸(B5)、吡哆醇(B6)、核黄素(B2)和硫胺素(B1),产生这些维生素的细菌可以占肠道微生物的40%-65%。近年来,逐渐有研究报导微生物在B族维生素生物合成中的作用。据估计,人类每日所需的维生素K一半都是由肠道微生物(如拟杆菌、双歧杆菌和肠球菌)提供的。值得注意的是,合成维生素K和水溶性B族维生素的酶在不同菌中有所不同。因为凝血素和骨钙蛋白合成都需要维生素K,因此其在凝血和骨骼生成中起着关键作用。此外,维生素K可以通过激活Gla蛋白来调节NF-κB/Nrf2通路,从而影响T2D中的血管炎症。此外,B族维生素还具有转录调节作用。例如,HCS-sGC-PKG通路调节生物素合成,Nrf2调节吡哆醇、钴胺素和泛酸合成,BDR4结合亚甲基四氢叶酸脱氢酶、环水解酶和甲酰四氢叶酸合成酶1(MTHFD1)等调节叶酸合成,GPR109调节尼克酸合成,DNA甲基化影响核黄素,p53影响硫胺素合成等。
胆汁酸(BAs)是由肝脏中的胆固醇合成的两亲性类固醇,一般称为初级胆汁酸。初级胆汁酸在小肠内被重吸收,结肠中的微生物对其进行结构修饰,形成次级胆汁酸。这一过程主要由7α/β-去羟基化酶介导。最近一项研究表明微生物的基因组结构变异与人类血浆胆汁酸相关,但这些结构变异的功能大部分仍然是未知的。结构变异(SVs)发生在细菌基因组上的高度可变区域,由于近来宏基因组测序技术的发展,鉴定到的SV主要包括存在/缺失变异和拷贝数变异。在过去的几十年中,已经发现了BAs许多重要功能,比如胆汁形成、促进肠道脂质和脂溶性维生素的吸收、维持胆固醇稳态以及在小肠中发挥抗菌作用。此外,已经证明BAs可以起到激素的作用,参与调控葡萄糖、脂质和能量代谢,调节免疫功能和细胞增殖,控制解毒反应等。BAs参与的反应主要通过激活核受体介导,即法尼类X受体(FXR)、维生素D受体(VDR)、PXR、雄烷受体(CAR)以及膜结合受体,如武田G蛋白偶联受体5(TGR5)和鞘氨醇-1-磷酸受体2(S1PR2)。重要的是,不同类型的初级和次级BAs由于结构不同,在经典功能和信号通路中发挥的作用不尽相同。这似乎是具有生理学意义的,因为在很多人群队列研究中都发现了不同个体间血浆BA浓度和组成的显著差异与肝脏脂肪含量、脂肪肝疾病、T2D以及多种血脂参数相关。
花青素是一类含有酚类结构的黄酮类化合物,广泛分布于植物液泡中,其颜色具有PH依赖性。花青素因其潜在的健康益处而被人熟知,它可以预防各种疾病,包括心血管疾病、癌症和神经退行性疾病,以及改善视觉和大脑功能。天然存在与食物中的常见花青素有花葵素、矢车菊素、花翠素、芍药色素、牵牛花色素和锦葵花素等。值得注意的是,花青素的益生元效应依赖微生物的调节。例如,肠道微生物分解矢车菊素-3-葡萄糖苷(C3G)产生酚类化合物,包括原儿茶酸、香草酸、间苯二醛和阿魏酸,从而激活Nrf2 、MAPK和NF-κB通路影响肠道中的氧化应激和炎症反应。微生物代谢花青素的产物鞣酸(GA)可以通过增加内皮型一氧化氮合酶(eNOS)的磷酸化来增加一氧化氮(NO)的水平。GA还抑制血管紧张素- I转换酶(ACE),降低血压。
植物雌激素是植物的非甾体次生代谢产物,具有独特的双酚类结构,包括多种类别的化合物,如二苯乙烯、香豆雌酚、异黄酮、鞣花丹宁和木酚素。植物雌激素广泛存在于我们的日常饮食中,具有各种物理化学和生物作用,包括抗氧化、抗菌、消炎、抗癌和保护心脏。与花青素相似,植物雌激素更倾向与雌激素受体结合。然而,通过肠道微生物酶促反应转化的植物雌激素变体生物活性显著增强。肠道微生物可以将植物雌激素转化为其他分子,如雌马酚、肠内酯和肠二醇。雌马酚可与在大脑多个区域都表达的核雌激素受体(ERs)结合,促进小脑发育。肠内酯和肠二醇都能缓解被脂多糖激活的外周血淋巴细胞的作用,进而导致抑制性-κB (I-κB)的降解和NF-κB的激活,从而产生TNF-α。
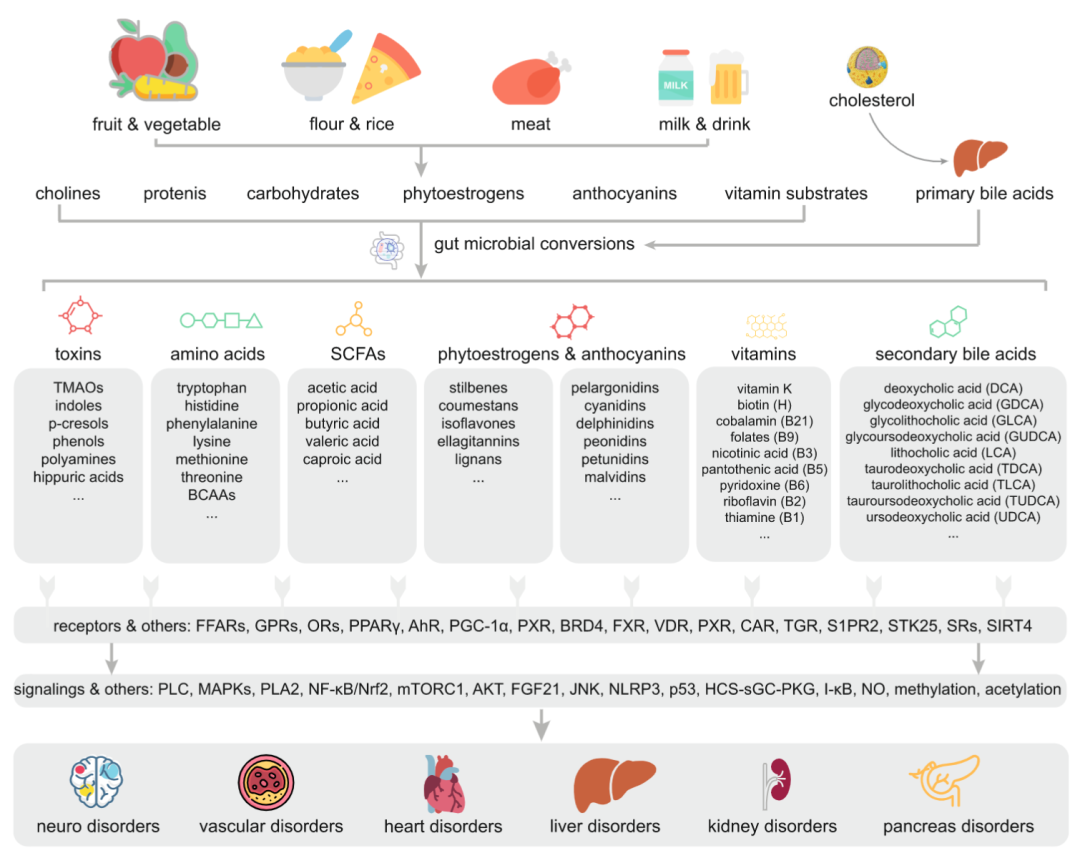
图2:影响人类健康的肠道微生物代谢物
肠道微生物参与了一系列具有生物活性的代谢物合成和转化,这些代谢物可以作为底物和信号分子,参与人类生理功能或者诱发复杂疾病。微生物相关代谢分子主要包括短链脂肪酸(SCFAs)、氨基酸(AAs)、维生素、胆汁酸(BAs)、毒素、花青素以及植物学雌激素等
药物互作
肠道微生物可以通过与药物的双向互作影响人类健康和疾病(图3)。一方面,许多肠道细菌在维持宿主代谢健康中发挥重要作用,抗生素可以通过一系列机制杀死这些细菌。例如,青霉素可以攻击细菌细胞壁阻止其合成肽聚糖,而肽聚糖是细菌在人体内存活所需的细胞壁强度主要来源。喹诺酮类药物通过靶向结合DNA螺旋酶(这种酶能够解开DNA双链进行复制)以防止细菌的增殖。四环素阻止关键分子结合到核糖体上的特定位点,从而阻止无性生殖。抗结核病的抗生素属于利福平类,都是通过抑制RNA的合成来发挥作用。
相反,常用的非抗生素药物也可受肠道微生物的影响,通过酶促反应改变其利用度、生物活性或毒性。最近一项体外试验,评估了来自人类肠道的76株细菌菌株(代表了主要细菌分类群中的68种)代谢271种药物的能力。这些药物是根据分子结构或对身体的影响等因素选择的。研究显示,有176种药物能够被至少一种菌株引起代谢变化,从而导致药物种活性分子水平的降低。这些结果都表明了大部分药物都能被微生物修饰/代谢,因此,通过对个体肠菌进行分离并测试其对药物的灭活作用将有助于推动个性化选药。

图3:微生物与药物的相互作用
肠道微生物可以通过与药物的双向互作影响人类健康和疾病。一方面,许多肠道细菌在维持宿主代谢健康中发挥重要作用,抗生素可以通过一系列机制杀死这些细菌。另一方面,常用的非抗生素药物可受肠道微生物的影响,通过酶促反应改变其利用度、生物活性或毒性
表征微生物功能的工具
基于下一代高通量测序技术,人们开发了一系列生物信息学工具来解码微生物DNA序列并预测其功能。一般来说,这些工具根据其原理可以分为三类,即基于分类标记基因的间接预测、基于基因同源性的直接预测和基于序列相似性的从头预测。
基于分类标记基因的间接预测
基于扩增子的标记基因(如16S rRNA)测序是评估和比较样本内或样本间微生物群落结构的有力工具。然而,由于这种技术序列信息仅来自特定的基因组区域,因此对肠道微生物功能的了解有限。不过,研究人员通常可以从已培养的物种中推断出未培养物种的功能,因为位于一个进化支的物种携带相同的核心基因。因此,一个物种基因组编码的功能可以基于相邻物种和注释良好的基因组而被部分预测。基于这个理论,研究人员已经开发出了基于分类信息学的肠道微生物组功能组分分析工具PICRUSt2、Tax4Fun2、BugBase和Piphillin等(表1)。
PICRUSt2利用一种基于IMG的扩展祖先状态重建算法对现有基因家族进行预测,然后通过加权法对这些基因家族进行组合,从而估计宏基因组的组成。Tax4Fun2依赖Ref100NR对最近邻居进行识别,进一步通过归一化和线性组合生成KEGG结果。Piphillin使用全局最近邻匹配算法生成独立于任何提议系统发育树的OTU丰度表,进一步结合最新的KEGG来分析功能组分。BugBase基于先验知识及系统发育树的方法,通过16S预测微生物表型(如耐氧量、革兰氏染色和致病潜能)。上述工具的预测能力主要依赖于可获得的基因组的功能信息,最近在基于宏基因组组装的基因组构建方面的进展则会大大提高功能预测的准确性。
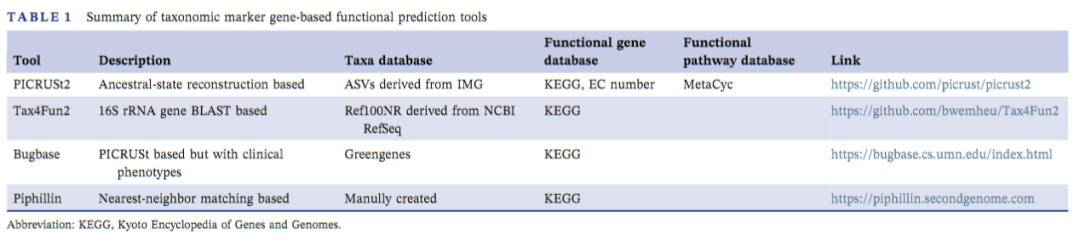
表1:基于分类标记基因预测工具总结
基于同源基因的直接预测
和基于分类标记基因的间接预测相比,鸟枪宏基因组/宏转录组测序(MGS)产生的大量测序reads能够覆盖整个基因组,这些reads通过和注释良好的数据库直接比对即可对肠道微生物功能进行更精准的预测。基于MGS进行功能预测的工具主要包括HUMAnN3 、MEGAN 、ShotMAP和 gutSMASH(表2)。HUMAnN3基于UniRef生成物种水平的基因丰度,并进一步将其分配到MetaCyc通路。MEGAN基于SEED、eggNOG和KEGG来描述微生物功能。ShotMAP将reads预测为开放阅读框(ORFs),进一步搜索SFams蛋白家族数据库。GutSMASH基于KnownClusterBlast和ClusterBlast数据库进行分类解析,挖掘重要的代谢基因簇,这些基因簇负责人体肠道微生物中各种代谢产物的生物合成。
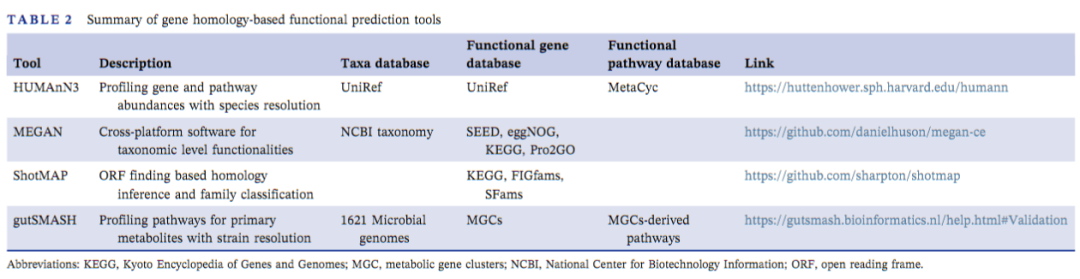
表2:基于同源基因预测工具总结
基于序列的从头预测
目前的微生物基因组注释方法都是基于序列相似性原理,与已有的数据库进行比较,比如UniRef、MetaCyc、SEED、eggNOG和KEGG。目前为止,只有接近60%的微生物基因组可以通过基于同源基因方法被注释,接近一半的微生物功能仍然是未解之谜。此外,因为从头突变(单核苷酸变异, SNVs)导致的遗传多样性迅速增加,而遗传多样性可以调控基因表达和功能。值得注意的是,现代分子生物学研究的一个普遍观点是一段基因序列定义了基因产物的结构,而这个结构反过来又决定了一个特定的功能。换句话说,即使两个基因序列相似性高达99%,但由于剩下1%的变异引起的结构差异,它们的功能也可能完全不同。因此,基于最终产物的结构(如蛋白质结构)来预测微生物基因的功能是一种很有前景的方法。最近,已经开发出一款机器学习的方法AlphaFold,在不知道蛋白质同源结构的情况下,其预测也能达到原子级别的精度。AlphaFold融合了有关蛋白质结构的物理和生物学知识,并将多序列比对设计在深度学习算法中。它没有强加已知的蛋白质生物物理学规则在其中,也没有模拟蛋白质折叠的物理过程。相反,AlphaFold从多次预测蛋白质结构的尝试中不断学习改进。因此,它可能会以一种全新的方式席卷微生物功能预测领域。
然而,在利用鸟枪法宏基因组测序数据来解码物种水平的微生物功能之前,还有很关键的一步是要对短reads的进行分箱。分箱基于是否依赖参考基因组主要分为两类。基于参考数据的方法是使用bowtie2等工具直接将reads比对到已有的微生物数据库,因此它的主要缺点是无法识别未知微生物基因组。不依赖参考数据的方法是一种无监督的方法,在没有任何参考数据库下将重叠群直接分箱。近年来,人们也评估了宏基因组分箱的各种工具,发现大多数基因组分箱工具虽然在独特菌株上表现良好,但在重构常见菌株上,所有基因组分箱工具仍然还面临重大挑战。这可能是因为这些常见的菌株具有相似的基因组,不易区分。尽管如此,长读长测序的发展将极大帮助从头分箱。
验证微生物功能的技术
在计算机方面,已经通过关联理论确定了数百种可能对人类健康与疾病产生影响的微生物。然而,对关注的肠道微生物进行预测的功能缺乏验证。因此,利用最先进的技术,如培养组学、基因编辑、新的研究模型以及多组学技术,可以进一步加强我们对其功能的理解,并开发完全基于肠道微生物组的个性化药物(图4)。
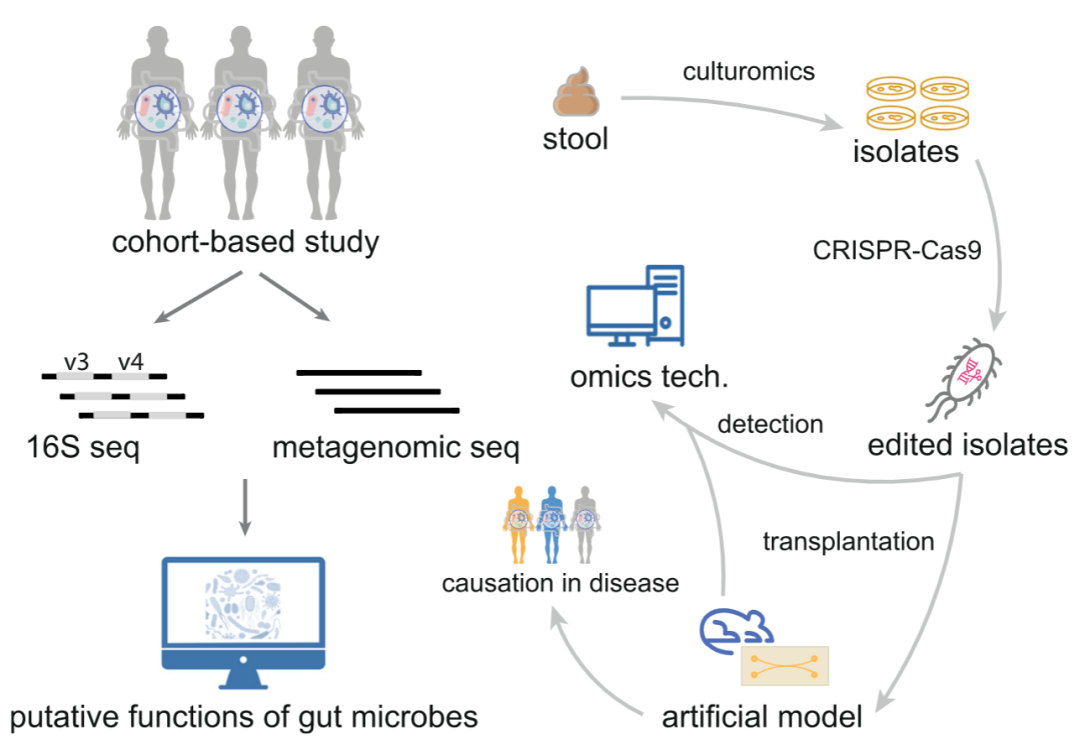
图4:利用先进技术进行微生物功能验证
在人类队列研究中,已经发现多种肠道微生物与各种疾病有关。虽然可以通过各种生物信息学工具预测肠道微生物的潜在功能,但缺乏功能验证。因此,利用最先进的技术,如培养组学、基因编辑、新的研究模型以及多组学技术,可以进一步加强我们对其功能的理解,并开发基于肠道微生物的个性化药物
培养组学
对肠道微生物组的测序表明,肠道中的大多数细菌仍未经过培养,揭示了特定肠道微生物功能的重要性。然而,该技术可能与DNA提取程序、生信工具、以及少数微生物种群的偏倚相关。因此,研究人员开发出培养组学来培养和鉴定人类肠道中的未知细菌,从而进行直接的功能验证和临床应用。培养组学是一种利用多种培养条件、质谱和测序来鉴定细菌种类的培养方法。培养组学的第一步是提供多种培养条件,促进分离自人类肠道的细菌生长。促进这些少数菌种的生长主要得益于培养基的改良。接下来,利用质谱技术快速鉴定微生物种类,该技术主要是分离蛋白质谱与最新的数据库进行比较。在此之后,利用16S和全基因组测序,通过和从人类身上恢复的微生物基因组进行比较来确认新的分类群。利用培养组学已经分离出数千种细菌菌株,其中很大一部分都被认为是新的物种/菌株。这些都允许我们在机制研究中,结合体外和动物模型或临床应用(如微生物群移植和CRISPR-Cas9基因组编辑),直接验证关联理论所作的功能假设。
基因编辑
研究表明,肠道微生物基因组中普遍存在结构变异。此外,基于全局宏基因组样本的SNP水平系统发育分析表明,物种间存在显著的遗传变异。在同一微生物菌株基因组中的变异可能导致其功能上有所不同。为了了解基因组变异的作用,可以利用CRISPR-Cas9等基因编辑工具修改微生物分离株的基因组,从而研究微生物的功能遗传调控机制。
人源化动物模型和类器官芯片模型
由于伦理问题,人类不能直接参与到肠道微生物在人类健康与疾病中功能的验证试验中。因此,新的体内和体外模型涌现出来,如人源化动物模型和类器官芯片技术,成为下一代研究疾病和药物模型。人性化动物模型是通过编辑动物基因组或诱导外部变化而获得的具有类人表型的动物模型。例如,通过CRISPR-Cas9基因组编辑技术敲掉Cyp2c70基因,可获得一个具有人类胆汁酸含量的小鼠模型,从而更好地揭示肠道微生物对BA代谢和CVD的影响。在器官芯片模型中,人诱导多能干细胞可以分化获得不同的组织和细胞类型。特别是像肠道芯片和肝脏芯片这样的模型对于研究微生物-肠道和宿主-微生物的相互代谢作用是非常有意义的。
多组学技术
除了通过基因组来研究微生物的功能,还可以通过生成各种类型的组学数据集,如代谢组学、蛋白质组学和转录组学,进一步将它们从分类关联到潜在功能都与肠道微生物组联系起来,加强我们对肠道微生物在复杂疾病中重要性的理解。然而,蛋白质组学和代谢组学的发展仍有很大挑战,阻碍了对肠道微生物功能的进一步探索。传统的靶向方法可以精确地识别和定量单个代谢产物或蛋白质,但其通量较低且成本较高,不太适用于大型队列研究。通过对MS-MS进行改进,非靶向方法可以在单次注射后分析数千个分子,然而,功能注释和量化仍然是这种方法的瓶颈。尽管在十多年前就发布了代谢物鉴定的公共指南,但一直没有一个推荐的标准。一个有效的解决方法就是为特定的代谢产物和蛋白质种类制定有针对性的提取/鉴定方案。此外,对酶功能的理解也在加深我们对未知代谢物和蛋白质的认识。代谢反应的知识将促进更强大识别工具的发展。对于转录组学,一个主要挑战就是微生物RNA的分离,因为粪便样本比较复杂,很难获得高质量的微生物RNA。然而,单细菌测序技术的发展可能是一个更好的解决方案。
未来展望
目前,微生物组的研究主要集中在微生物类群和功能基因丰度方面。但依靠测序数据产生的肠道微生物丰度并不能真正反映人类肠道中微生物的真实密度(绝对丰度)。如果样品之间微生物的负荷差异很大,相对分析将会阻碍微生物组特征与定量数据(如代谢物浓度)之间的关联。因此,近年来出现了利用流式细胞仪将微生物细胞计数,随后与粪便微生物组测序数据相结合的微生物组定量分析方法,该方法在评估个体内部和个体之间的微生物变化方面有更大的潜力。然而,缺点是该方法取决于实验技巧,且单一的测量并不能很好地估计平衡丰度。此外,该方法非常昂贵且耗时,并不适用于大队列研究。因此,该技术还有很大的进步空间。
除了非常重要的微生物基因拷贝数变异之外,研究肠道微生物基因组的变异也是一个重要的方向。与人类基因组一样,微生物DNA序列中的SNPs、SV(如插入和缺失)、移动遗传元件(如噬菌体和转座元件)对微生物功能也至关重要,并与人类疾病相关。在过去的几十年里,微生物功能的遗传调控研究主要集中在有限的已知基因上。多组学技术可将生理测量或代谢物浓度与微生物全基因组系统关联(例如,SNPs和sv),揭示肠道微生物在宿主健康和疾病中的新功能,且其分辨率比微生物丰度高得多,然而,相关的研究极其缺乏。然而,在生物信息学(如在上述章节中讨论过的基因组分箱问题)和统计学(例如,微生物基因组中数百万个SNP增加了统计测试的数量,这将对检测能力产生负面影响)方面仍存在许多挑战。在不久的将来,长读长测序和常见变异研究可能是一个潜在的探索方向。
结 论
肠道菌群对宿主的影响主要是通过微生物毒力因子、代谢分子以及与药物的双向互作来介导的。我们归纳了肠道微生物在人类健康和疾病中的潜在功能,并总结了相关的分子机制。此外,我们还介绍了相关的生物信息学工具,可以用于解码微生物基因组的功能,研究人员可以根据其特定目的去使用它们。最后,我们介绍了培养组学、基因组编辑、多组学技术以及功能验证新模型的重要性,并阐述了通过调节肠道微生物来改善人类健康的可能性。
引文格式:
Wang, Yifeng, Dong, Quanbin, Hu, Shixian, Zou, Huayiyang, Wu, Tingting, Shi, Jing, Zhang, Haifeng, et al. 2022. “ Decoding Microbial Genomes to Understand their Functional Roles in Human Complex Diseases.” iMeta. e14. https://doi.org/10.1002/imt2.14
作者简介

王屹丰(第一作者)
● 博士,助理研究员,江苏省人民医院心血管内科
● 博士毕业于南京医科大学流行病与卫生统计学系,主要从事心血管疾病相关危险因素研究。研究成果发表在 Nature Cell Biology, American Journal of Human Genetics,Frontiers in Cell and Developmental Biology, Gene等杂志,目前国家自然科学基金青年项目1项。

孔祥清(通讯作者)
● 教授,主任医师,医学博士,博士生导师。国家百千万人才、国家有突出贡献中青年专家、江苏省“333工程”第一层次培养对象、江苏省有突出贡献中青年专家等
● 现任苏州市立医院党委书记,江苏省人民医院心内科主任。同时兼任国家标准化心血管与代谢疾病中心主任、中华医学会心血管病学分会常委、中华医学会心血管病学分会结构心脏病学组组长、国家心血管病专家委员会委员、中国医师协会心血管内科分会转化医学副主委、美国心血管造影和介入学会委员(FSCAI)、欧洲心脏病学学会委员(FESC)等职。获首届全国创新争先奖、国家技术发明二等奖1项,教育部科技进步一等奖1项,江苏省科技进步二等奖1项,江苏省科技进步三等奖1项,江苏省卫生厅新技术引进一等奖3项等

陈连民(通讯作者)
● 博士,教授,南京医科大学/江苏省人民医院海外引进人才(学术带头人)
● 荷兰格罗宁根大学博士(cum laude)及博士后,主要从事肠道微生物在宿主心血管健康中的作用研究。至今共发表论文 40 余篇, 包括Cell(封面文章),Nature,Nature Medicine,Nature Genetics,Circulation Research(封面文章),Nature Communications 及 Cell Reports (封面文章)等
更多推荐
(▼ 点击跳转)
iMeta | 北大陈峰组综述口腔微生物组的标准化研究:从技术驱动到假说驱动
iMeta | 电子科大林昊组开发蛋白质赖氨酸乳酸化位点预测工具DeepKla

iMeta | 南昌大学丁霞等-水产养殖系统对中华鳖微生物组和肠道代谢组的影响
iMeta | 华中科大宁康组综述用于蛋白质结构预测的宏基因组定量分析
iMeta | 中科院李小方等膳食甘草促进小鼠镉解毒并调节肠道菌群代谢
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
iMeta | 中科院生态中心邓晔组发布微生物组网络分析平台iNAP
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号
iMeta
责任编辑
微微
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集



















 466
466

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









