






生成PDB文件通常需要根据蛋白质的氨基酸序列、原子坐标(xyz)、占有率(occupancy)和B因子(B-factor)等信息来格式化输出。
示例代码,展示如何根据这些信息生成PDB文件。
RES_NAMES = [
'ALA','ARG','ASN','ASP','CYS',
'GLN','GLU','GLY','HIS','ILE',
'LEU','LYS','MET','PHE','PRO',
'SER','THR','TRP','TYR','VAL'
]
RES_NAMES_1 = 'ARNDCQEGHILKMFPSTWYV'
#to1letter = {aaa:a for a,aaa in zip(RES_NAMES_1,RES_NAMES)}
#to3letter = {a:aaa for a,aaa in zip(RES_NAMES_1,RES_NAMES)}
ATOM_NAMES = [
("N", "CA", "C", "O", "CB"), # ala
("N", "CA", "C", "O", "CB", "CG", "CD", "NE", "CZ", "NH1", "NH2"), # arg
("N", "CA", "C", "O", "CB", "CG", "OD1", "ND2"), # asn
("N", "CA", "C", "O", "CB", "CG", "OD1", "OD2"), # asp
("N", "CA", "C", "O", "CB", "SG"), # cys
("N", "CA", "C", "O", "CB", "CG", "CD", "OE1", "NE2"), # gln
("N", "CA", "C", "O", "CB", "CG", "CD", "OE1", "OE2"), # glu
("N", "CA", "C", "O"), # gly
("N", "CA", "C", "O", "CB", "CG", "ND1", "CD2", "CE1", "NE2"), # his
("N", "CA", "C", "O", "CB", "CG1", "CG2", "CD1"), # ile
("N", "CA", "C", "O", "CB", "CG", "CD1", "CD2"), # leu
("N", "CA", "C", "O", "CB", "CG", "CD", "CE", "NZ"), # lys
("N", "CA", "C", "O", "CB", "CG", "SD", "CE"), # met
("N", "CA", "C", "O", "CB", "CG", "CD1", "CD2", "CE1", "CE2", "CZ"), # phe
("N", "CA", "C", "O", "CB", "CG", "CD"), # pro
("N", "CA", "C", "O", "CB", "OG"), # ser
("N", "CA", "C", "O", "CB", "OG1", "CG2"), # thr
("N", "CA", "C", "O", "CB", "CG", "CD1", "CD2", "CE2", "CE3", "NE1", "CZ2", "CZ3", "CH2"), # trp
("N", "CA", "C", "O", "CB", "CG", "CD1", "CD2", "CE1", "CE2", "CZ", "OH"), # tyr
("N", "CA", "C", "O", "CB", "CG1", "CG2") # val
]
# 氨基酸中原子的编号到具体的原子名称的映射:('A', 0): ('ALA', 'N')
idx2ra = {(RES_NAMES_1[i],j):(RES_NAMES[i],a) for i in range(20) for j,a in enumerate(ATOM_NAMES[i])}
def writepdb(f, xyz, seq, bfac=None):
f.seek(0)
ctr = 1
seq = str(seq)
L = len(seq)
if bfac is None:
bfac = np.zeros((L))
idx = []
for i in range(L):
for j,xyz_ij in enumerate(xyz[i]):
key = (seq[i],j)
if key not in idx2ra.keys():
continue
# 有些氨基酸结构缺失,xyz_ij shape为(3,)
if np.isnan(xyz_ij).sum()>0:
continue
r,a = idx2ra[key]
f.write ("%-6s%5s %4s %3s %s%4d %8.3f%8.3f%8.3f%6.2f%6.2f\n"%(
"ATOM", ctr, a, r,
"A", i+1, xyz_ij[0], xyz_ij[1], xyz_ij[2],
1.0, bfac[i,j] ) )
if a == 'CA':
idx.append(i)
ctr += 1
#f.close()
f.flush()
return np.array(idx)
chainA = chains['A']
print(f"chainA['xyz']维度:{chainA['xyz'].shape}")
print(f"chainA['seq']长度:{len(chainA['seq'])}")
print(f"chainA['bfac']维度:{chainA['bfac'].shape}")
with open("temp_A.pdb",'w') as f:
writepdb(f, chainA['xyz'],chainA['seq'],chainA['bfac'])
#print(chainA['xyz'][0,1].shape)
#print(chainA['xyz'].shape)
#print(chainA['seq'])
#print(chainA['bfac'])
#print(idx2ra.keys())
chainA的数据可以从mmcif结构文件中单独提取出A链的数据。
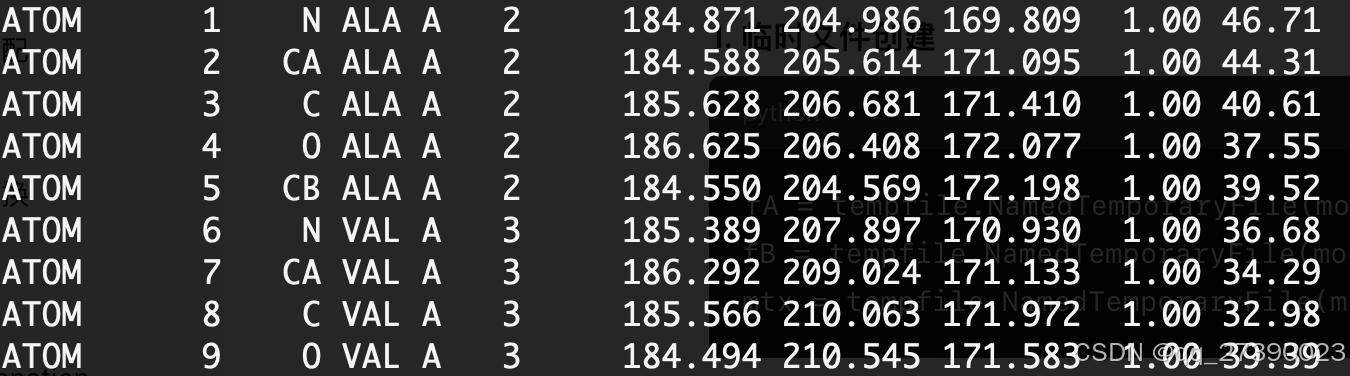
temp_A.pdb文件内容:

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









