






深圳杯作为竞赛时长一个月,上半年度数模竞赛中难度最大的竞赛,会被各种省级竞赛、高校作为选拔赛进行选拔。本文为了能够帮助大家快速的上手该题目,将从给出D题超详细解题思路。
对于本题目,我们首先需要对给出的数据进行必要的了解。
原题目给出数据说明:
以《附件1:不同人数的STR图谱数据》中第一个数据的Sample File为例,说明贡献者编号和贡献者比例。“A02_RD14-0003-40_41-1;4-M3S30-0.075IP-Q4.0_001.5sec.fsa”中40_41-1;4表示该样本由《附件3:各个贡献者对应的基因型数据》中Sample ID为40、41的两位贡献者的样本混合,混合比例为1:4。
基因座(marker)是染色体上一个特定的物理位置,等位基因(allele)是同一基因座上不同表现形式的DNA序列。在STR图谱中,每一个主峰代表一个等位基因,其size表示该STR等位基因的DNA片段长度,不同size对应不同的等位基因,height是峰高,反映该等位基因的DNA量,可用于判断样本是否为混合样本。
具体而言,对于
附件1:不同人数的STR图谱数据 展示51种不同人数组合方案的816种结果数据。人数组合方案为2、3、4、5,混合比例均为1:1
附件2:不同混合比例的STR图谱数据.xlsx 展示50种不同人数、混合比例组合方案的816种结果数据
指标含义(来自网络,真实性自行判断)
| 表格名称 | 具体含义 |
| Dye(染料) | 标记DNA片段的荧光染料 |
| Allele(等位基因 ) | 某个特定基因座(位置)上检测到的等位基因 |
| Size(大小) | 该基因座的等位基因对应的DNA片段的大小,单位通常是碱基对(bp) |
| Height(高度) | 该基因座上等位基因的峰值高度,通常反映了该等位基因在混合样本中的相对丰度或DNA量 |
| Marker | **STR基因座(短串联重复序列)**的名称 |
附件3:各个贡献者对应的基因型数据.xlsx 第1列 'Research ID',第16列 'AM'与后续处理相关性不大,不进行处理。表格中 第一行为基因Name 下面的每一行数据为每个人对应该基因信息,对应的每一个人每个基因有两个数据,使用,分隔,这是对应基因的位置编号。
我们首先对该数据进行可视化,下述可视化完整实现代码见文末(完全免费)
| 以下为python绘图 | 以下为matlab绘图 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

问题1:混合STR图谱分析中的贡献者人数判断
在多人犯罪案件中,法医DNA鉴定需要分析混合STR图谱。混合样本中包含多个人的DNA信息,问题的核心是准确地判断混合样本中参与贡献的DNA贡献者人数。识别贡献者人数的正确性是整个分析过程的基础,错误的贡献者人数判断将直接影响后续分析的准确性。
Y=f(x,z,t,a,j)
提取每个样本的Sample File和对应的STR图谱数据,包括基因座(Marker)、不同等位基因的大小(Size)和高度(Height)。对每个基因座的STR图谱进行分析,提取每个基因座的不同等位基因的大小和高度。对于每个样本,记录每个基因座的峰数(即等位基因的数量),以及这些峰的大小和高度。
对于每个样本的STR图谱,可以使用聚类算法(如K-means、DBSCAN等)对图谱中的峰进行分类,推测贡献者的数量。假设样本中有多个贡献者时,我们可以依据峰的分布情况(大小、数量、重叠情况)进行聚类,从而推测出贡献者的人数。聚类算法可参考文末给出数模中常见聚类算法
创新点:
l自动化特征提取:结合深度学习模型(如卷积神经网络)自动从STR图谱中提取峰值特征,进一步提高贡献者人数的识别准确
l多任务学习:同时学习贡献者人数和基因型推断,通过共享特征提高整体模型的效果。
l时间序列分析:对于长时间序列的STR数据,可以采用时间序列模型(如LSTM)进一步捕捉样本的动态变化。
| Python示例代码(实际运行代码需等待进一步更新) | matlab示例代码(实际运行代码需等待进一步更新) |
| import numpy as np import pandas as pd from sklearn.cluster import KMeans import matplotlib.pyplot as plt # 假设数据:每个基因座的峰值数据(Size和Height) # 数据结构:[Marker, Size, Height] data = [ ["D8S1179", 5.2, 200], ["D8S1179", 5.5, 150], ["D8S1179", 6.0, 100], ["D21S11", 7.2, 220], ["D21S11", 7.5, 180], ["D21S11", 8.0, 130], # 更多数据... ] # 将数据转为 DataFrame df = pd.DataFrame(data, columns=["Marker", "Size", "Height"]) # 提取不同的基因座 markers = df["Marker"].unique() # 对于每个基因座,提取对应的 Size 和 Height 并进行聚类分析 for marker in markers: marker_data = df[df["Marker"] == marker][["Size", "Height"]].values # 使用 K-means 聚类来推测贡献者人数 k = 2 # 假设贡献者人数为 2 kmeans = KMeans(n_clusters=k, random_state=0) kmeans.fit(marker_data) # 聚类结果 labels = kmeans.labels_ centers = kmeans.cluster_centers_ # 可视化聚类结果 plt.figure() plt.scatter(marker_data[:, 0], marker_data[:, 1], c=labels, cmap='viridis', s=50) plt.scatter(centers[:, 0], centers[:, 1], c='red', marker='x', s=200, label='Centroids') plt.title(f'Clustered Peaks for {marker}') plt.xlabel('Size') plt.ylabel('Height') plt.legend() plt.show() # 输出聚类结果 print(f"Cluster labels for {marker}: {labels}") print(f"Cluster centers for {marker}: {centers}") | % 数据结构:[Marker, Size, Height] data = [ "D8S1179", 5.2, 200; "D8S1179", 5.5, 150; "D8S1179", 6.0, 100; "D21S11", 7.2, 220; "D21S11", 7.5, 180; "D21S11", 8.0, 130; % 更多数据... ]; % 提取不同的基因座标 markers = unique(data(:, 1)); % 对于每个基因座,提取对应的Size和Height for i = 1:length(markers) marker_data = data(strcmp(data(:, 1), markers{i}), 2:3); % 聚类:K-means,用来识别贡献者 k = 2; % 假设贡献者人数为2 [idx, C] = kmeans(marker_data, k); % 可视化聚类结果 figure; scatter(marker_data(:, 1), marker_data(:, 2), 50, idx, 'filled'); title(['Clustered Peaks for ', markers{i}]); xlabel('Size'); ylabel('Height'); legend({'Contributor 1', 'Contributor 2'}); end |
问题2:贡献者比例判断
在分析混合STR图谱中的贡献者人数后,下一步是判断各贡献者在混合样本中的比例。贡献者比例的判断对于进一步推测基因型是非常重要的,尤其当贡献者的比例接近时,等位基因可能会重叠,导致误判基因型。对于该问题,数据包含了50种不同人数、混合比例组合方案的816种结果数据,需要根据这些数据建立一个模型,推断混合样本中各个贡献者的比例。
首先,需要处理给定的数据,确保数据格式一致并清理缺失值。数据包含不同混合比例的STR图谱信息,每个样本的基因座特征(Size 和 Height)。数据中包含贡献者比例信息,需要将比例与对应的图谱特征进行匹配。
提取每个基因座的峰值特征,包括每个等位基因的大小(Size)和高度(Height)。这些特征反映了混合样本中各个贡献者的DNA比例。可能还需要计算基因座之间的相关性,了解不同基因座上的DNA片段如何相互作用。
利用已知的混合比例数据来训练一个回归模型,推测混合样本中每个贡献者的比例。
l回归模型:可以使用多元线性回归、岭回归(Ridge Regression)、Lasso回归等方法,通过拟合混合样本中的峰值特征与已知贡献者比例之间的关系。
l支持向量回归(SVR):如果数据具有较强的非线性特征,可以使用支持向量回归模型来建立贡献者比例与STR图谱特征之间的映射关系。
创新点:
l自适应特征选择:可以根据数据的实际情况,自适应选择不同的基因座作为特征输入,优化模型的表现。
l深度学习:使用深度神经网络或卷积神经网络(CNN)进行特征提取和比例预测,自动学习图谱中的复杂模式。
l强化学习:可以通过强化学习来逐步调整模型预测的混合比例,优化模型在复杂情况下的预测准确性。
| Python示例代码(实际运行代码需等待进一步更新) | matlab示例代码(实际运行代码需等待进一步更新) |
| import numpy as np import pandas as pd from sklearn.linear_model import Ridge from sklearn.model_selection import train_test_split from sklearn.metrics import mean_squared_error import matplotlib.pyplot as plt # 假设数据:从附件2读取数据,包含基因座(Size 和 Height)及混合比例(Contributor Proportions) # 加载数据 data = pd.read_excel('附件2:不同混合比例的STR图谱数据.xlsx') # 假设数据结构如下: # data包含多个基因座的Size和Height列(比如 'Size1', 'Height1', 'Size2', 'Height2', ...) # 以及对应的贡献者比例列(比如 'Proportion_Contributor1', 'Proportion_Contributor2', ...) # 提取特征和标签 X = data[['Size1', 'Height1', 'Size2', 'Height2', 'Size3', 'Height3']] # 选择特征 y = data[['Proportion_Contributor1', 'Proportion_Contributor2']] # 目标:贡献者比例 # 数据分割:训练集与测试集 X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42) # 使用岭回归进行建模 ridge_reg = Ridge(alpha=1.0) ridge_reg.fit(X_train, y_train) # 预测测试集的贡献者比例 y_pred = ridge_reg.predict(X_test) # 计算模型的均方误差(MSE) mse = mean_squared_error(y_test, y_pred) print(f"Mean Squared Error: {mse}") # 可视化预测结果与实际值的比较 plt.scatter(y_test['Proportion_Contributor1'], y_pred[:, 0], label='Contributor 1') plt.scatter(y_test['Proportion_Contributor2'], y_pred[:, 1], label='Contributor 2') plt.plot([0, 1], [0, 1], color='red', linestyle='--') # 对角线 plt.xlabel('Actual Proportions') plt.ylabel('Predicted Proportions') plt.legend() plt.title('Actual vs Predicted Proportions') plt.show() | % 加载数据 data = readtable('附件2:不同混合比例的STR图谱数据.xlsx'); % 假设数据结构如下: % data包含多个基因座的Size和Height列(比如 'Size1', 'Height1', 'Size2', 'Height2', ...) % 以及对应的贡献者比例列(比如 'Proportion_Contributor1', 'Proportion_Contributor2', ...) % 提取特征和标签 X = data{:, {'Size1', 'Height1', 'Size2', 'Height2', 'Size3', 'Height3'}}; % 选择特征 y = data{:, {'Proportion_Contributor1', 'Proportion_Contributor2'}}; % 目标:贡献者比例 % 数据分割:训练集与测试集(80%训练,20%测试) cv = cvpartition(size(X, 1), 'HoldOut', 0.2); X_train = X(training(cv), :); y_train = y(training(cv), :); X_test = X(test(cv), :); y_test = y(test(cv), :); % 使用岭回归进行建模 lambda = 1; % 岭回归的正则化参数 B = ridge(y_train, X_train, lambda, 0); % 岭回归模型训练 % 预测测试集的贡献者比例 y_pred = X_test * B(2:end, :) + B(1, :); % 计算模型的均方误差(MSE) mse = mean((y_test - y_pred).^2, 'all'); fprintf('Mean Squared Error: %.4f\n', mse); % 可视化预测结果与实际值的比较 figure; scatter(y_test(:, 1), y_pred(:, 1), 'filled'); hold on; scatter(y_test(:, 2), y_pred(:, 2), 'filled'); plot([0, 1], [0, 1], 'r--'); % 对角线 xlabel('Actual Proportions'); ylabel('Predicted Proportions'); legend('Contributor 1', 'Contributor 2', 'Location', 'best'); title('Actual vs Predicted Proportions'); hold off; |
问题3:贡献者比例判断
根据附件1(混合STR图谱数据)和附件2(不同混合比例的STR图谱数据),以及附件3(各个贡献者的基因型数据),我们的任务是推断混合STR图谱中每个贡献者的基因型。基因型指的是在每个基因座上个体的等位基因组合。通过准确地推断出贡献者的基因型,可以为案件的分析提供更加精确的信息。
从附件1中提取混合STR图谱数据,包含各基因座(Marker)、等位基因大小(Size)、峰高(Height)等信息。从附件2中提取贡献者的比例和相关图谱数据。从附件3中提取已知贡献者的基因型数据。
利用已知贡献者的基因型数据作为训练数据,建立基因型与图谱特征之间的映射关系。使用监督学习方法(例如回归模型或分类模型),根据混合样本中的图谱特征推断出贡献者的基因型。可以通过最大似然估计(MLE)或贝叶斯推断方法,根据图谱中峰的大小和高度推断基因型。
创新点:
l深度学习方法:可以使用卷积神经网络(CNN)或循环神经网络(RNN)处理图谱数据,自动提取特征并进行基因型推断。
l多任务学习:在推断基因型的同时,结合贡献者比例信息进行联合训练,提升推断准确度。
l贝叶斯网络:使用贝叶斯网络模型将基因型与峰值信息建模,进行推断并评估每种基因型的可能性。
| Python示例代码(实际运行代码需等待进一步更新) | matlab示例代码(实际运行代码需等待进一步更新) |
| import numpy as np import pandas as pd from sklearn.svm import SVC from sklearn.model_selection import train_test_split from sklearn.metrics import accuracy_score import matplotlib.pyplot as plt # 假设数据:从附件1读取混合STR图谱数据 # 假设附件3提供已知贡献者基因型数据 # 数据结构:'Size1', 'Height1', 'Size2', 'Height2' 等,以及贡献者基因型 'Genotype_Contributor1', 'Genotype_Contributor2' # 加载数据 data = pd.read_excel('附件1:混合STR图谱数据.xlsx') # 提取特征和标签 X = data[['Size1', 'Height1', 'Size2', 'Height2', 'Size3', 'Height3']] # 选择特征 y = data[['Genotype_Contributor1', 'Genotype_Contributor2']] # 目标:基因型 # 数据分割:训练集与测试集 X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42) # 使用支持向量机(SVM)进行基因型推断 svm_model = SVC(kernel='linear', random_state=42) svm_model.fit(X_train, y_train) # 预测测试集的基因型 y_pred = svm_model.predict(X_test) # 计算准确率 accuracy = accuracy_score(y_test, y_pred) print(f'Accuracy: {accuracy * 100:.2f}%') # 可视化实际值与预测值的比较 plt.scatter(y_test['Genotype_Contributor1'], y_pred[:, 0], label='Contributor 1') plt.scatter(y_test['Genotype_Contributor2'], y_pred[:, 1], label='Contributor 2') plt.plot([0, 1], [0, 1], color='red', linestyle='--') # 对角线 plt.xlabel('Actual Genotype') plt.ylabel('Predicted Genotype') plt.legend() plt.title('Actual vs Predicted Genotype') plt.show() | import numpy as np import pandas as pd from sklearn.svm import SVC from sklearn.model_selection import train_test_split from sklearn.metrics import accuracy_score import matplotlib.pyplot as plt # 假设数据:从附件1读取混合STR图谱数据 # 假设附件3提供已知贡献者基因型数据 # 数据结构:'Size1', 'Height1', 'Size2', 'Height2' 等,以及贡献者基因型 'Genotype_Contributor1', 'Genotype_Contributor2' # 加载数据 data = pd.read_excel('附件1:混合STR图谱数据.xlsx') # 提取特征和标签 X = data[['Size1', 'Height1', 'Size2', 'Height2', 'Size3', 'Height3']] # 选择特征 y = data[['Genotype_Contributor1', 'Genotype_Contributor2']] # 目标:基因型 # 数据分割:训练集与测试集 X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42) # 使用支持向量机(SVM)进行基因型推断 svm_model = SVC(kernel='linear', random_state=42) svm_model.fit(X_train, y_train) # 预测测试集的基因型 y_pred = svm_model.predict(X_test) # 计算准确率 accuracy = accuracy_score(y_test, y_pred) print(f'Accuracy: {accuracy * 100:.2f}%') # 可视化实际值与预测值的比较 plt.scatter(y_test['Genotype_Contributor1'], y_pred[:, 0], label='Contributor 1') plt.scatter(y_test['Genotype_Contributor2'], y_pred[:, 1], label='Contributor 2') plt.plot([0, 1], [0, 1], color='red', linestyle='--') # 对角线 plt.xlabel('Actual Genotype') plt.ylabel('Predicted Genotype') plt.legend() plt.title('Actual vs Predicted Genotype') plt.show() |
问题4:贡献者比例判断
在处理混合STR图谱数据时,去噪技术能够有效减少噪声干扰,提高数据的质量。然而,去噪过程可能会带来一些影响,例如对信号的细节和峰形的丢失。去噪可能导致一些有用信息的损失,尤其是在信号非常微弱或重叠的情况下。此时,我们需要评估去噪后的图谱数据如何影响后续分析,如贡献者人数的识别、比例推测和基因型的推断。
对比去噪前后的STR图谱数据,分析去噪处理是否有效保留了有用的信息,避免过度去噪。主要评估去噪后是否出现了峰值丢失、峰形变化或其他影响结果的现象。
影响评估指标:
l信噪比(SNR):去噪后,信噪比是否提高?信噪比越高,去噪效果越好,数据质量更高。
l均方误差(MSE):计算去噪前后混合样本的MSE,评估去噪的影响。若去噪后的误差显著增大,则可能表明去噪过度。
l准确度和F1分数:分析去噪后的贡献者人数识别、比例推测和基因型推断的准确性。
| Python示例代码(实际运行代码需等待进一步更新) | matlab示例代码(实际运行代码需等待进一步更新) |
| import numpy as np import matplotlib.pyplot as plt from sklearn.metrics import mean_squared_error import pywt from keras.models import Sequential from keras.layers import Conv1D, MaxPooling1D, Flatten, Dense # 假设数据:混合STR图谱(带噪声和去噪后信号) t = np.linspace(0, 10, 500) signal = np.sin(t) + np.random.normal(0, 0.5, 500) # 带噪声的信号 # 使用小波变换去噪 coeffs = pywt.wavedec(signal, 'db1', level=2) coeffs[1:] = [np.zeros_like(c) for c in coeffs[1:]] # 去除高频噪声 denoised_signal = pywt.waverec(coeffs, 'db1') # 计算去噪前后的均方误差 mse_before = mean_squared_error(signal, np.sin(t)) # 与真实信号(假设真实信号是sin(t))对比 mse_after = mean_squared_error(denoised_signal, np.sin(t)) # 可视化去噪前后的信号 plt.figure() plt.plot(t, signal, label='Noisy Signal') plt.plot(t, denoised_signal, label='Denoised Signal', linewidth=2) plt.legend() plt.title('Signal Denoising Comparison') plt.xlabel('Time') plt.ylabel('Amplitude') plt.show() print(f'MSE Before Denoising: {mse_before}') print(f'MSE After Denoising: {mse_after}') # 使用CNN进行去噪(仅示例) model = Sequential([ Conv1D(32, 3, activation='relu', input_shape=(signal.shape[0], 1)), MaxPooling1D(2), Conv1D(64, 3, activation='relu'), MaxPooling1D(2), Flatten(), Dense(1) ]) # 数据预处理 signal_scaled = signal.reshape(-1, 1, 1) model.compile(optimizer='adam', loss='mse') model.fit(signal_scaled, signal_scaled, epochs=10) # 使用CNN进行去噪 denoised_signal_cnn = model.predict(signal_scaled).flatten() # 可视化CNN去噪结果 plt.figure() plt.plot(t, signal, label='Noisy Signal') plt.plot(t, denoised_signal_cnn, label='Denoised Signal (CNN)', linewidth=2) plt.legend() plt.title('Signal Denoising Using CNN') plt.show() | % 假设数据:混合STR图谱(带噪声和去噪后信号) t = linspace(0, 10, 500); signal = sin(t) + randn(1, 500) * 0.5; % 带噪声的信号 % 使用小波变换去噪 [coeffs, levels] = wavedec(signal, 2, 'db1'); coeffs(2:end) = 0; % 去除高频噪声 denoised_signal = waverec(coeffs, levels, 'db1'); % 计算去噪前后的均方误差 mse_before = immse(signal, sin(t)); % 与真实信号(假设真实信号是sin(t))对比 mse_after = immse(denoised_signal, sin(t)); % 可视化去噪前后的信号 figure; plot(t, signal, 'r', 'DisplayName', 'Noisy Signal'); hold on; plot(t, denoised_signal, 'b', 'DisplayName', 'Denoised Signal'); legend; title('Signal Denoising Comparison'); xlabel('Time'); ylabel('Amplitude'); hold off; disp(['MSE Before Denoising: ', num2str(mse_before)]); disp(['MSE After Denoising: ', num2str(mse_after)]); % 使用卷积神经网络(CNN)进行去噪(示例) autoenc = trainAutoencoder(signal', 10, 'MaxEpochs', 50, 'HiddenLayerSize', 25, 'TrainingAlgorithm', 'trainscg'); denoised_signal_autoenc = predict(autoenc, signal'); % 可视化自编码器去噪结果 figure; plot(t, signal, 'r', 'DisplayName', 'Noisy Signal'); hold on; plot(t, denoised_signal_autoenc', 'g', 'DisplayName', 'Denoised Signal (Autoencoder)'); legend; title('Signal Denoising Using Autoencoder'); xlabel('Time'); ylabel('Amplitude'); hold off; |
数据预处理代码
| Python代码 | matlabd代码 |
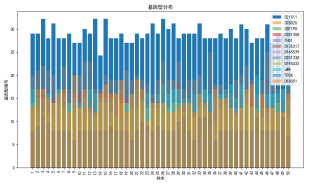
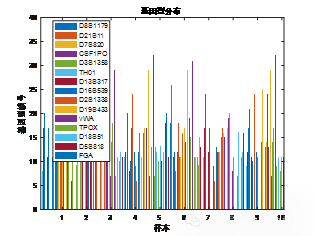
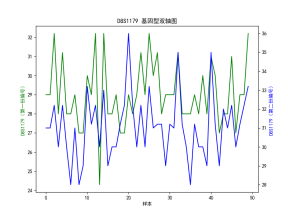
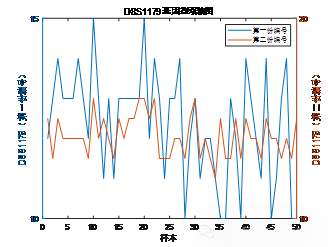
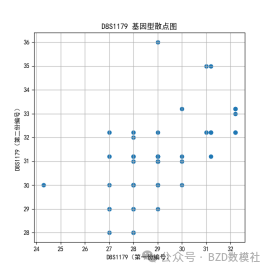
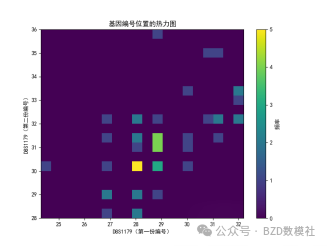
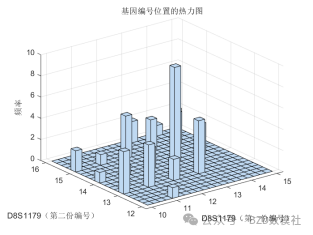
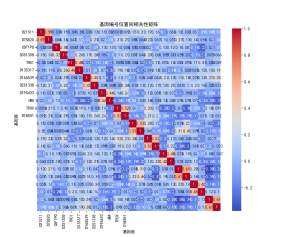
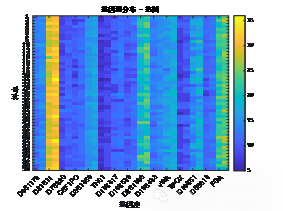
| # 读取数据 import pandas as pd import numpy as np import matplotlib.pyplot as plt import seaborn as sns plt.rcParams['font.sans-serif'] = ['SimHei'] # 使用SimHei字体 plt.rcParams['axes.unicode_minus'] = False # 正确显示负号 # 读取Excel数据 data = pd.read_excel('附件3:各个贡献者对应的基因型数据.xlsx') # 去除不需要的列(第1列 'Research ID', 第2列 'Sample ID', 第16列 'AM') data_clean = data.iloc[:, [2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 16, 17]] # 保留第3到第15列以及第17和第18列 # 获取基因名称(从第三列开始是基因座数据) gene_names = data_clean.columns[1:-2] # 获取基因名称(去掉最后两列,因为它们不是基因数据) # 获取样本数量 num_samples = len(data) num_genes = len(gene_names) # 初始化基因型矩阵,存储每个样本在各个基因座上的两个位置编号 genotypes = np.zeros((num_samples, 2 * num_genes)) # 每个基因两个编号 # 提取每个样本在各个基因座上的基因型 for i in range(num_genes): for j in range(num_samples): genotype_str = str(data_clean.iloc[j, i + 1]) # 获取基因型数据(例如 '12,15') genotype = list(map(float, genotype_str.split(','))) # 转换为浮动列表并分割 genotypes[j, 2 * i] = genotype[0] # 第一份编号 genotypes[j, 2 * i + 1] = genotype[1] # 第二份编号 # 绘制条形图 plt.figure(figsize=(10, 6)) plt.bar(np.arange(num_samples), genotypes[:, 0], label=gene_names[0]) # 只绘制第一个基因 for i in range(1, num_genes): plt.bar(np.arange(num_samples), genotypes[:, 2 * i], label=gene_names[i], alpha=0.5) plt.title('基因型分布') plt.xlabel('样本') plt.ylabel('基因型编号') plt.xticks(np.arange(num_samples), data['Sample ID'], rotation=90) # 使用 'Sample ID' 作为 X 轴标签 plt.legend(loc='best') plt.tight_layout() plt.show() # 绘制热图 plt.figure(figsize=(12, 8)) sns.heatmap(genotypes, cmap='viridis', xticklabels=gene_names, yticklabels=data['Sample ID'], cbar=True) plt.title('基因型分布 - 热图') plt.xlabel('基因座') plt.ylabel('样本') plt.show() # 选择两个基因座进行散点图绘制 gene_1 = genotypes[:, 0] # 第一个基因座的第一份编号 gene_2 = genotypes[:, 1] # 第一个基因座的第二份编号 # 绘制散点图 plt.figure(figsize=(6, 6)) plt.scatter(gene_1, gene_2) plt.title('D8S1179 基因型散点图') plt.xlabel('D8S1179(第一份编号)') plt.ylabel('D8S1179(第二份编号)') plt.grid(True) plt.show() # 计算每个基因编号位置之间的皮尔逊相关系数 correlation_matrix = np.corrcoef(genotypes.T) # 绘制相关性矩阵热图 plt.figure(figsize=(10, 8)) sns.heatmap(correlation_matrix, cmap='coolwarm', annot=True, xticklabels=gene_names, yticklabels=gene_names) plt.title('基因编号位置间相关性矩阵') plt.xlabel('基因座') plt.ylabel('基因座') plt.show() # 假设我们对第一个基因座 D8S1179 绘制双轴图 gene_1 = genotypes[:, 0] # 第一个编号 gene_2 = genotypes[:, 1] # 第二个编号 fig, ax1 = plt.subplots(figsize=(8, 6)) ax2 = ax1.twinx() ax1.plot(np.arange(num_samples), gene_1, 'g-') ax2.plot(np.arange(num_samples), gene_2, 'b-') ax1.set_xlabel('样本') ax1.set_ylabel('D8S1179(第一份编号)', color='g') ax2.set_ylabel('D8S1179(第二份编号)', color='b') plt.title('D8S1179 基因型双轴图') plt.show() # 绘制热力图 plt.figure(figsize=(8, 6)) plt.hist2d(gene_1, gene_2, bins=20, cmap='viridis') plt.title('基因编号位置的热力图') plt.xlabel('D8S1179(第一份编号)') plt.ylabel('D8S1179(第二份编号)') plt.colorbar(label='频率') plt.show() | % 读取数据 data = readtable('附件3:各个贡献者对应的基因型数据.xlsx'); % 去除不需要的列(第1列 'Research ID', 第2列 'Sample ID', 第16列 'AM') data_clean = data(:, [3:15, 17, 18]); % 保留第3到第15列以及第17和第18列 % 获取基因名称(从第三列开始是基因座数据) gene_names = data_clean.Properties.VariableNames; % 获取基因名称(列名) % 获取样本数量 num_samples = height(data); num_genes = length(gene_names); % 初始化基因型矩阵,存储每个样本在各个基因座上的两个位置编号 genotypes = zeros(num_samples, 2 * num_genes); % 每个基因两个编号 % 提取每个样本在各个基因座上的基因型 for i = 1:num_genes for j = 1:num_samples % 获取单元格内容并处理基因型字符串 genotype_str = data_clean{j, i}; % 获取基因型数据(例如 '12,15') genotype = str2double(strsplit(char(genotype_str), ',')); % 转换为字符向量并分割 genotypes(j, 2*i-1) = genotype(1); % 第一份编号 genotypes(j, 2*i) = genotype(2); % 第二份编号 end end % 绘制条形图 figure; bar(genotypes); title('基因型分布'); xlabel('样本'); ylabel('基因型编号'); xticklabels(data.SampleID); % 使用 'Sample ID' 作为 X 轴标签 legend(gene_names, 'Location', 'Best'); % 绘制热图 figure; imagesc(genotypes); colorbar; title('基因型分布 - 热图'); xlabel('基因座'); ylabel('样本'); xticks(1:2:2*num_genes); xticklabels(gene_names); yticks(1:num_samples); yticklabels(data.SampleID); % 使用 'Sample ID' 作为 Y 轴标签 % 选择两个基因座进行散点图绘制 gene_1 = genotypes(:, 1); % 第一个基因座的第一份编号 gene_2 = genotypes(:, 2); % 第一个基因座的第二份编号 % 绘制散点图 figure; scatter(gene_1, gene_2); title('D8S1179 基因型散点图'); xlabel('D8S1179(第一份编号)'); ylabel('D8S1179(第二份编号)'); % 计算每个基因编号位置之间的皮尔逊相关系数 correlation_matrix = corr(genotypes); % 绘制相关性矩阵热图 figure; imagesc(correlation_matrix); colorbar; title('基因编号位置间相关性矩阵'); xlabel('基因座'); ylabel('基因座'); xticks(1:2:2*num_genes); xticklabels(gene_names); % 基因座名称 yticks(1:2:2*num_genes); yticklabels(gene_names); % 基因座名称 % 假设我们对第一个基因座 D8S1179 绘制双轴图 gene_1 = genotypes(:, 1); % 第一个编号 gene_2 = genotypes(:, 2); % 第二个编号 figure; [ax, h1, h2] = plotyy(1:num_samples, gene_1, 1:num_samples, gene_2); title('D8S1179 基因型双轴图'); xlabel('样本'); ylabel(ax(1), 'D8S1179(第一份编号)'); ylabel(ax(2), 'D8S1179(第二份编号)'); legend([h1, h2], {'第一份编号', '第二份编号'}); % 计算每个基因编号的分布 gene_1 = genotypes(:, 1); % 第一个编号 gene_2 = genotypes(:, 2); % 第二个编号 % 绘制热力图 figure; hist3([gene_1, gene_2], [20, 20]); % [20, 20] 是每个轴的分组数 title('基因编号位置的热力图'); xlabel('D8S1179(第一份编号)'); ylabel('D8S1179(第二份编号)'); zlabel('频率'); |

















 1144
1144

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









