




一、概述
本教程提供了使用 MITOS 进行线粒体基因组注释的方法,包括网页版 MITOS 和本地版 MITOS 的使用流程,并且对注释结构进行解释。MITOS 的注释结果中有 RNA 的二级结构图可以用于后续的美化。首先推荐大家优先使用网页版的 MITOS 进行注释,因为本地版需要一定的代码基础,如果课题组有服务器条件并且有大量的基因组需要注释可以考虑使用本地版本的 MITOS。
二、视频教学
三、网页版 MITOS 注释流程
优先推荐使用网页版 MITOS,操作简单。MITOS 的官网网址是:HTTP://mitos.bioinf.uni-leipzig.de/index.py,目前官网已经不提供注释服务,我们需要进入网页中提供的镜像网页进行注释。
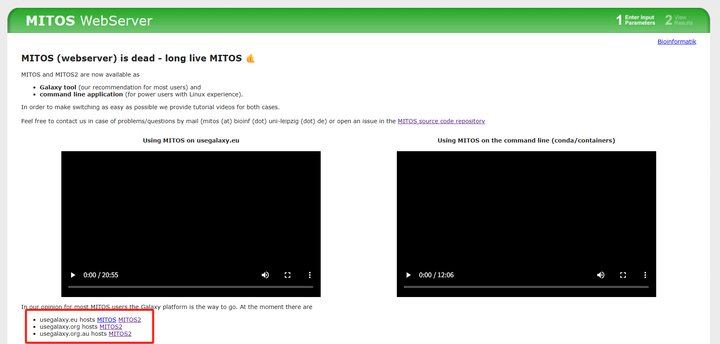
MITOS官网界面,红色方框是镜像网页的超链接
推荐使用 MITOS2 的注释服务,个别物种或者参考文献中可能会提到使用 MITOS 1.0 版本的复现研究过程中才使用 MITOS 1.0。有时 MITOS 网页打不开,可以使用官方提供的链接:
-
usegalaxy.eu hosts MITOS(1.0 版本,不推荐) MITOS2
-
usegalaxy.org hosts MITOS2
-
usegalaxy.org.au hosts MITOS2
可以根据自身网络条件进行选择!你也可以选择国内的银河服务器镜像:MITOS2(国内镜像),接下来以 usegalaxy.eu 镜像为例,其他的镜像使用方法一致。
3.1 序列导入
在注释开始之前我们需要有组装完整的线粒体基因组 Fasta 序列,并且文件中必须有且仅有一条序列,例如我们如果有一个组装好的线粒体基因组 fasta 格式的序列:
>s1 [organism=Alectoris magna] ACCCTCCACAGGCAAAAGGAGCAGGTATCAGGCACACCCAATGTTTAGCCCAAGACGCCTTGCTAAG CCACACCCCCACGGGTATTCAGCAGTAATTAACCTTAAGCAATAAGTGCAAACTTGACTTAGCCATA GCAATACTAGGGTTGGTAAATCTTGTGCCAGCCACCGCGGTCATACAAGAAACCCAAATTAATAGCC ATCCGGCGTAAAGAGTGGCCACATGTTATCCGCATTAACTAAGATCAAAACGAAACTGAGCTGTCAT AAGCCCAAGACTCACCTAAGCCCGACCTAAAAACCATCTTAGTTCCCACGACCAATTTAAACCCACG ... ACAGGCAAAAGGAGCAGGTATCAGGCACACCCAATGTTTAGCCCAAGACGCCTTGCTAAGCCACACC CCCACGGGTATTCAGCAGTAATTAACCTTAAGCAATAAGTGCAAACTTGACTTAGCCATAGCAATAC
我们可以将其添加到网页中
第一步:点击浏览本地文件夹
第二步:点击上传
第三步:选择本地文件
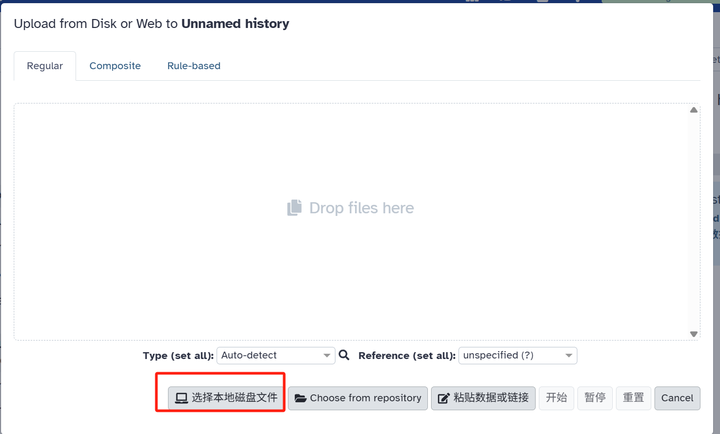
第四步:指定格式
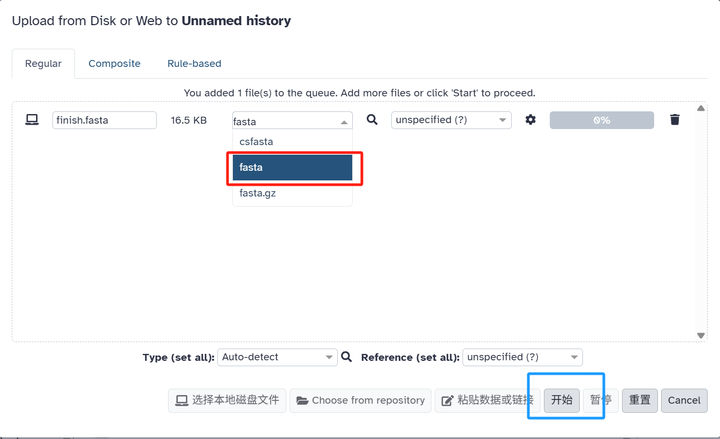
第五步:点击开始即可导入,点击取消可以关闭窗口
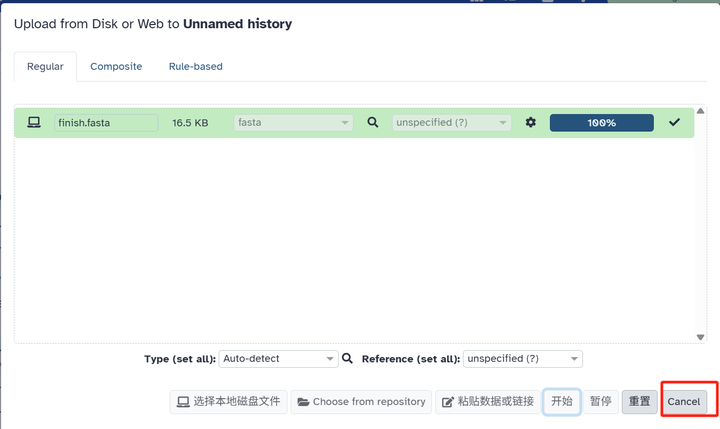
第六步:选择我们上传的序列文件
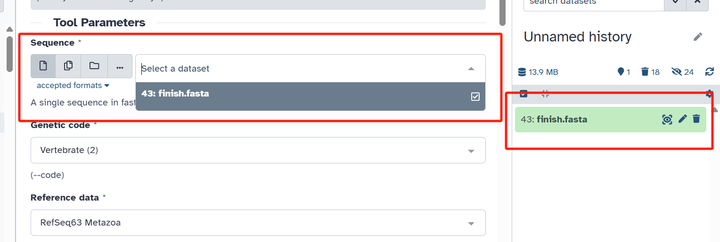
3.2 密码子表选择
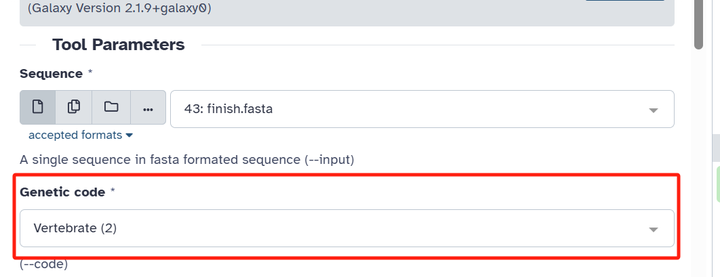
在密码子表处选择物种线粒体对应的密码子表,注意:无论什么物种,只要是核基因都是严格遵循标准密码子表的,只有线粒体和质体基因可能有其他遵循,由于本教程是面向线粒体基因组的,所以需要按照实际调整。脊椎动物线粒体是第 2 套密码子表,无脊椎动物线粒体是第 5 套密码子表,更多物种的密码子表遵循规则可以参考 NCBI 提供的网页:HTTPS://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi。

3.3 参考基因组选择
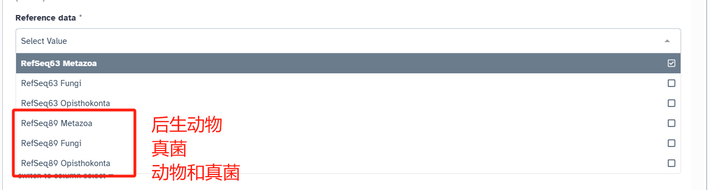
如果没有特殊需求,请选择 89 版本参考基因组!Metazoa 表示后生动物,Fungi 表示真菌,Opisthokonta 表示动物和真菌。如果需要复现文章可能会有使用 63 版本的机会。
3.4 指定序列是否是线性的
对于线粒体基因组来说不是线性,默认 No 即可。
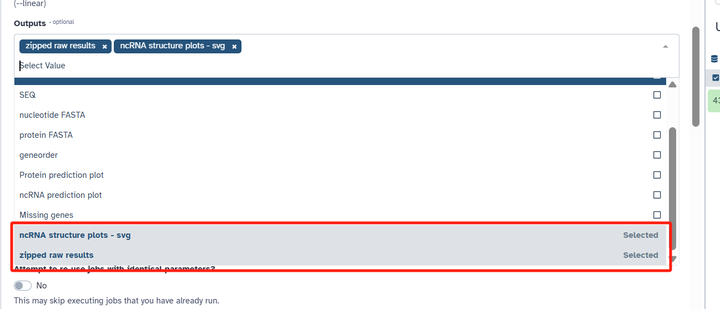
3.5 导出结果指定
因为每个导出文件都需要在后台跑一次程序,所以为了节省时间只需要导出 zipped raw results 即可输出全部内容,但是有时候压缩包里包括的内容不完整,比如 tRNA 的二级结构图是最容易缺失的,所以我们可以顺便把 ncRNA structure plots -svg 勾选上。
3.6 参数设置
我们只需要关注特征选择即可,其他参数正常保持默认即可。

特征把能勾选的都勾选上
3.7 运行工具
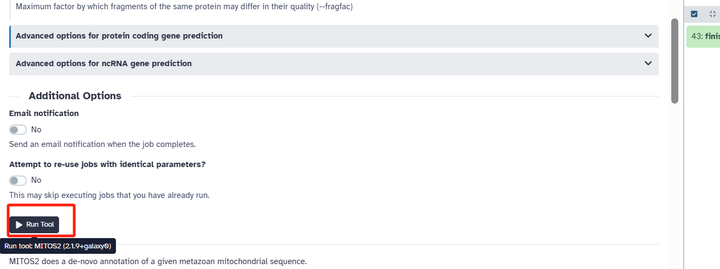
点击 Run 就开始注释了!
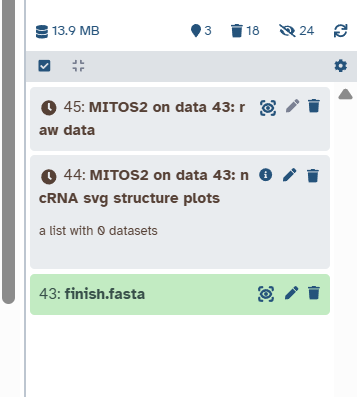
之后工作窗口就显示我们的注释任务了!我们勾选几个输出格式就会有几个程序在跑,所以建议不要点太多的输出类型。
3.8 下载结果
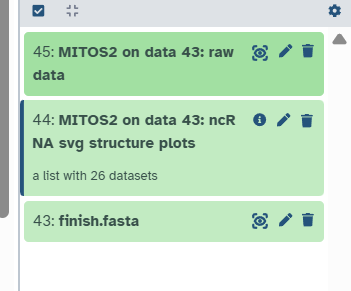
绿色表示可以下载结果了!我只下载压缩包文件,因为里边什么结果都有(不出意外的话)。
如果显示压缩包无法打开,重新下载一遍即可
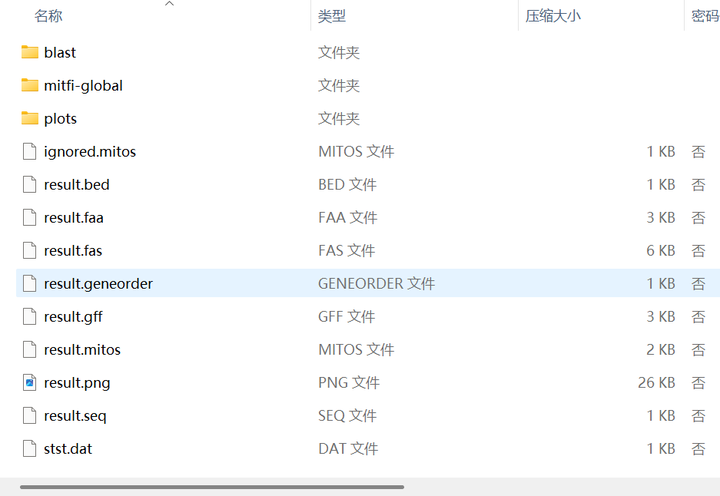
下载完成之后有以下内容:
四、结果解释
在 zipped raw results 的结果中包含所有的输出文件。
4.1 blast 文件夹
包含了蛋白编码基因预测过程中核苷酸和氨基酸的 blast 比对的结果。
4.2 mitfi-global 文件夹
包含了 ncRNA 编码基因预测过程中的结果。
4.3 plots 文件夹(重要)
里边包含了非编码 RNA 的二级结构图的 SVG 以及 PS 格式。其中 SVG 版本的图片使用我的美化脚本可以组图美化。

4.4 其他文件
ignored.mitos:不重要;
result.bed:bed 格式注释文件;
result.faa:注释出的氨基酸序列;
result.fas:注释出的基因序列;
result.geneorder:基因排布;
result.gff:gff 格式注释文件;
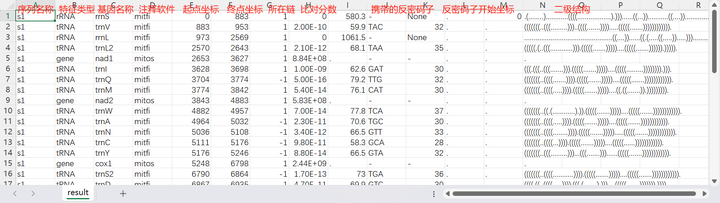
result.mitos:mitos 格式注释文件,里边包含的信息最多,其中起止位置坐标从 0 开始,实际位置坐标应该是表格中的数字加 1;
result.png:基因排布图;
result.seq:tbl 格式注释文件;
stst.dat:不重要。
五、本地服务器版 MITOS 注释流程(以 MITOS2 为例,选做)
本地版的源代码托管于:HTTPS://GitLab.com/Bernt/MITOS
5.1 下载安装
安装方法1:使用conda建立mitos环境,并进入mitos环境
conda create --strict-channel-priority -c conda-forge -c bioconda -n mitos2 "mitos>=2"
conda activate mitos2
安装方法2:使用mamba建立mitos环境,并进入mitos环境(推荐)
mamba create --strict-channel-priority -c conda-forge -c bioconda -n mitos2 "mitos>=2"
conda activate mitos2
安装方法3:使用pip安装(不推荐)
pip install mitos
安装报错cmsearch未安装需要运行:可能需要root权限,建议联系管理员
apt install infernal
运行:runmitos.py –help如果看到参数说明,即为安装成功!
5.2 主要参数
主要参数:
-i FILE, --input FILE 输入的基因组文件;
-C CODE, --code CODE 需要使用的密码子表(填写序号);
-o OUTDIR, --outdir OUTDIR 指定输出文件夹(需要预先创建);
-r REFSEQVER, --refseqver REFSEQVER 参考基因组文件夹相对-refdir 的路径;
-R REFDIR, --refdir REFDIR 有些鸡肋,必须指定;
--zip 指定可以输出压缩包。
其他参数可参照帮助信息。
5.3 参考基因组下载
本地版注释软件需要自行下载参考基因组并解压,参考基因组托管在 zenodo 数据库中(MITOS, MITOS2),由于网络原因可能打不开,可以尝试使用下面的指令下载。类比 3.3 的说明,推荐使用 89 版本的参考基因组 89m 表示后生动物,89f 表示真菌,89o 表示动物和真菌,推荐需要哪个下载哪个。
使用wget下载所需要的参考基因组:
wget https://zenodo.org/records/4284483/files/refseq89m.tar.bz2
wget https://zenodo.org/records/4284483/files/refseq89f.tar.bz2
wget https://zenodo.org/records/4284483/files/refseq89o.tar.bz2
wget https://zenodo.org/records/4284483/files/refseq63f.tar.bz2
wget https://zenodo.org/records/4284483/files/refseq63m.tar.bz2
wget https://zenodo.org/records/4284483/files/refseq63o.tar.bz2
解压:
(单个解压)tar jxvf refseq89m.tar.bz2
(批量解压)for i in `ls |grep .tar.bz2`;do tar jxvf $i;done
如果下载不了我也整理了这些参考基因组,可以自行下载:
MITOS2参考基因组,提取码:wqej![]() https://pan.baidu.com/s/1tCHWNf0fZkLdFUJ3KP8Rqg?pwd=wqej如果链接失效请私信。
https://pan.baidu.com/s/1tCHWNf0fZkLdFUJ3KP8Rqg?pwd=wqej如果链接失效请私信。
5.4 示例代码
conda activate mitos2
wget https://zenodo.org/records/4284483/files/refseq89m.tar.bz2
tar jxvf refseq89m.tar.bz2
mkdir output
nohup runmitos.py --input input.fasta --code 2 --outdir ./output --refdir '/' --refseqver /mnt/sdb/xuehao/mitostest/refseq89m&
例子中,input.fasta 是线粒体基因组,--code 是指定第几套密码子表,例子中是脊椎动物。
六、注意事项
Mitos 只适用于动物和真菌线粒体基因组注释,植物质体(线粒体/叶绿体)基因组应使用其他工具(如 GeSeq);
Mitos 注释可以提供较为准确的注释结果,但由于物种差异等原因造成注释有偏差,应当对比多个近缘物种的基因组结构进行人工注释修正;
可以结合近缘物种的基因组结构对基因注释的数目进行评估,如果注释数目少于其他物种,在保证基因组组装完整的前提下,调整阈值参数以得到更多的候选基因。

















 59
59

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









