










OmicsTools的R语言分析环境配置教程效果示意图
软件介绍
我开发了一款本地电脑无限使用的零代码生信数据分析作软图神器电脑软件OmicsTools,欢迎大家使用OmicsTools进行生物医学科研数据分析和作图,该软件件能让大家在不需要任何编程和代码编写的基础上,分析次数没有限制,可以无限使用,让您在自己的电脑上快速进行大量的生信分析和加速大家的科学研究。
软件下载
我开发的本地电脑无限使用无限分析作图的生信零代码一键分析电脑软件神器OmicsTools 软件在github上的zihaoxingstudy1/OmicsTools仓库中,大家可以下载安装OmicsTools进行各种生信分析和可视化作图。
更新的软件下载
OmicsTools软件是一直都在更新,而且是永久更新的,而且更新非常频繁,一直在为大家增加更多的生信分析功能,一般每隔两周都会更新一版OmicsTools并添加更多新的功能进来。
OmicsTools的更新和优化包括实时推送的日常小更新优化和版本整体的增量大更新,平时用户反馈参数的改动和新特性和一些问题我都会快速进行优化,用户只需要把软件进程关掉,重启一下,就能实时享受到最新的功能和解决掉反馈的故障问题,大版本的更新我每隔两周左右发一个新版本的安装包。
我更新的在本地电脑无限使用无限分析作图的生信零代码一键分析电脑软件神器OmicsTools新版本软件都可以到我的zihaoxingstudy1/OmicsTools github仓库中下载,也可以到我的生信交流qq群931846486的群文件中下载到OmicsTools分析软件,大家可以一直下载安装更新的OmicsTools软件进行各种生信分析和可视化作图。
如何能试用OmicsTools?
OmicsTools试用链接(https://s.click.taobao.com/Vlrmumt) (可以试用七天)
OmicsTools依赖的R语言分析环境配置
R语言+Rtools+大量的R包的总文件体积非常庞大,可能快到10个G了,打包的软件体积不能太大,这样不利于用户下载软件,所以,我的OmicsTools软件包体积是200多MB,内部没有包含R语言完整的分析环境,但是我都给每个OmicsTools会员用户发了一份R语言+Rtools+所有被我编译好的直接拷贝到自己电脑上就能开箱即用的R包下载网盘链接,还有详细的b站视频教学链接,大家按照我发的网盘文件和我录制的b站教学视频都能很容易的把R语言的分析配置好。
在这里,我在跟大家提一下R语言分析环境配置时需要注意的一些细节操作。
特别提示(这是大家安装配置时最需要注意的事情):
如何快速一次性配置好OmicsTools的R语言生信分析环境?
- 不要直接用我百度网盘里文件下载解压缩后的目录和名字,你应该在你的电脑上跟我视频和这篇文章教程里演示的那样创建一模一样的目录和路径,如果这些目录和路径在你的电脑里还不存在,就在你电脑里创建同样的目录名称的路径。
- 切记,这些目录的名称和路径一定要跟我的视频和这篇文章教程截图里的完全一模一样就可以一次性配置成功了。
OmicsTools安装配置的b站教学视频
生信科研数据分析作图神器OmicsTools电脑软件安装使用教程
这个安装配置视频将的非常详细,大家一定要把这个视频从头到尾完整看一遍,大家完整的安装这个视频的教学来操作,基本上都可以把OmicsTools分析环境配置好的。
所有生信分析的R包我都 编译打包了一份,大家 下载到自已电脑上就可 以开箱即用了,能解决 各种R包安装失败问题
这个补充答疑的视频大家也认真看看,讲解了如何试用我网盘里的所有开箱即用的R包以及最新分享的R包, Rlibs目录该如何配置,以及怎么解决Seurat报错问题,对于配置OmicsTools构建完整的生信分析R环境也是非常重要的。
R语言+Rtools+git整套软件的下载安装
软件下载
可以从我发的百度网盘链接中下载这一整套软件,或者从官网上下载,但是要下载R-4.3.2这个版本。

压缩包中有这些软件

R语言安装
R语言必须要安装到D:\omics_tools\R\R-4.3.2这个目录下,安装好的示意图如下:

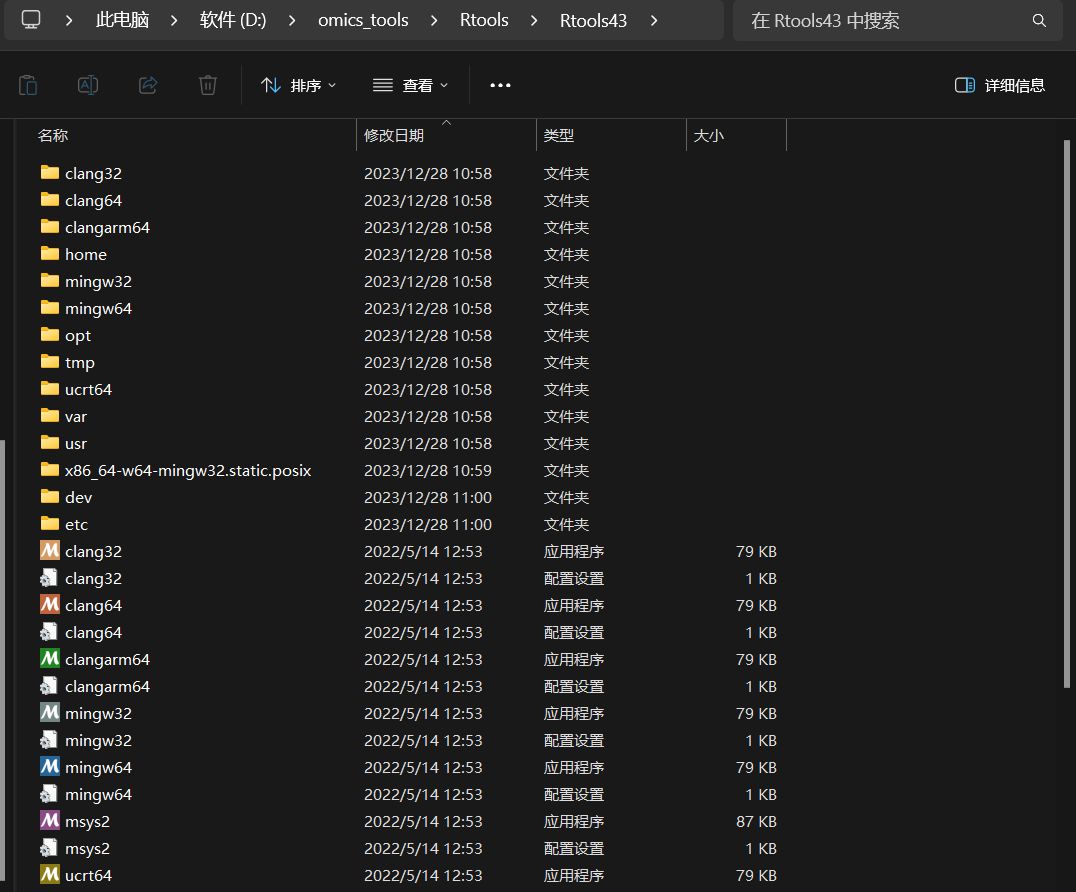
Rtools安装
Rtools在windows系统中R包安装等常见非常有用,大家需要安装到你们的电脑中。Rtools要安装到D:\omics_tools\Rtools\Rtools43这个目录下。
安装好的示意图如下:
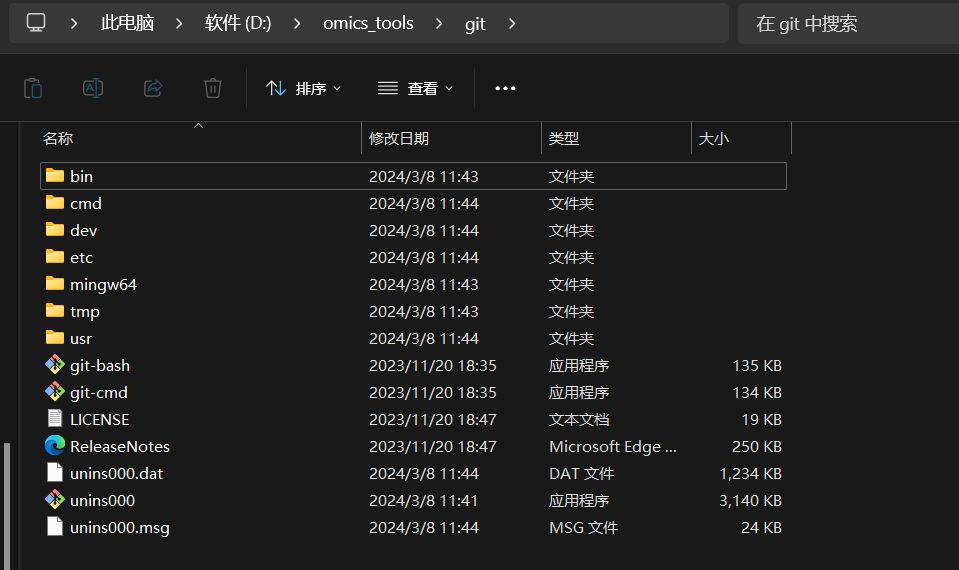
Git软件安装
Git软件在windows系统中也很有用,从github上下载文件或R包都很有用,需要安装一下,git软件要安装到D:\omics_tools\git这个目录下。
安装好的示意图如下:
把安装好的这些软件的启动路径添加到用户环境变量
这些安装好的软件都需要添加到环境变量中才能再终端中正常的使用R语言进行各种生信分析和科研作图。
添加到环境变量的操作示意图如下:


需要注意的是,这个添加环境变量的路径都要跟我截图的这个路径是一模一样的,这样就是能配置好的,另外,D:\omics_tools中的omics_tools目录名称是小写的,大家不要命名成大写的了。
下载解压所有开箱即用的R包
R包的网盘文件
我给大家发了很多开箱即用的R包下载网盘链接,这是最全的R包的网盘文件,其中Rlibs_part_a_c.rar, Rlibs_part_d_o.rar,Rlibs_part_p_t.rar,Rlibs_part_u_z.rar是我把D:\omics_tools\Rlibs\R-4.3.2原来所有的R包名称从a-z所有R包拆分了4份压缩后上传到了百度网盘,因为百度网盘不让上传太大的压缩包,同时add_dir.rar是我日常有更新或新增的一些R包后补充上传到百度网盘的压缩包。
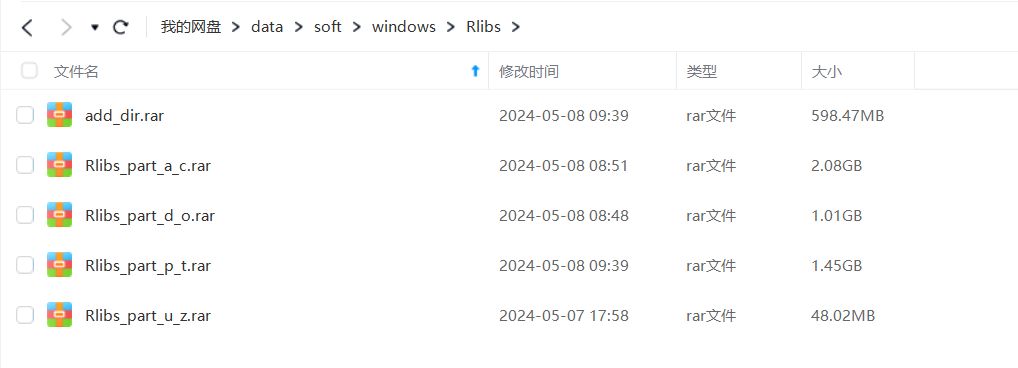
全部R包的解压方式和解压移动好的效果示意图
大家把上面的这五个R包下载到本地电脑后,先把这五个R包都解压一下,因为add_dir.rar这个压缩包的R包时最新更新的一些R包,所以add_dir.rar一定要放在最后解压,如果add_dir.rar第一个解压,后面旧版本有重名的R包就会替换add_dir.rar中新版本的R包,R包的版本就又变旧了,大家解压的顺序要是先解压Rlibs_part_a_c.rar,Rlibs_part_d_o.rar;Rlibs_part_p_t.rar;Rlibs_part_u_z.rar这四个压缩后,然后再解压add_dir.rar这个压缩包,如果add_dir.rar压缩包中有跟前四个压缩包中重名的R包,就选择‘全部替换’, 用add_dir.rar中最新的R包替换掉前面四个part中重名的旧R包,就能达到R包定期更新的目的了。
然后一定要进入解压后的目录里并把这些R包目录中所有的一个个R包全部都复制移动到D:\omics_tools\Rlibs\R-4.3.2这同一个目录下。所有的R包移动到D:\omics_tools\Rlibs\R-4.3.2里面后,这些R包就能开箱即用,不用大家再辛苦安装各种R包了,所有R包解压并移动好的示意图如下:

替换修改掉R语言的Rprofile.site配置文件
把R语言自带的Rprofile.site文件用我发给大家的Rprofile.site文件替换一下,就能适配omics_tools来用R语言做各种生信分析了。
R语言配置文件Rprofile.site文件替换操作如下:
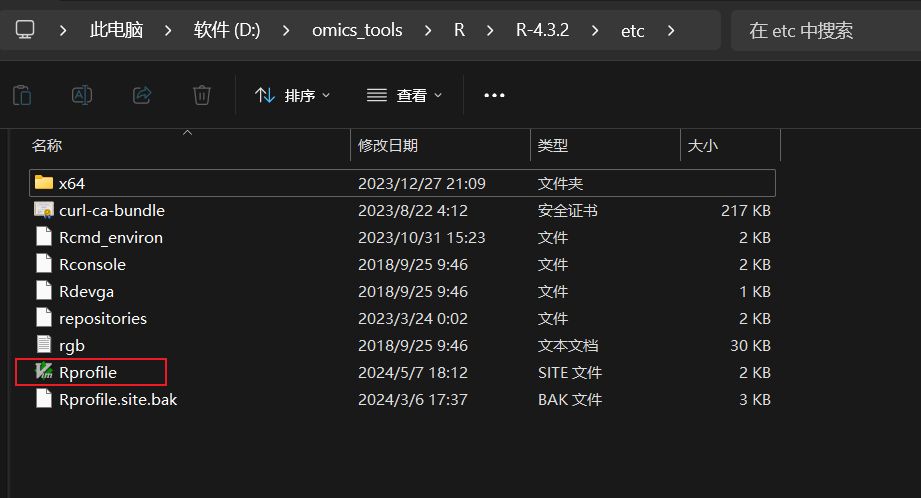
D:\omics_tools\R\R-4.3.2\etc目录下有个原始的Rprofile.site配置文件,大家把这个配置文件替用发你们的网盘链接里的Rprofile.site文件替换一个,R的分析环境就配置好了。

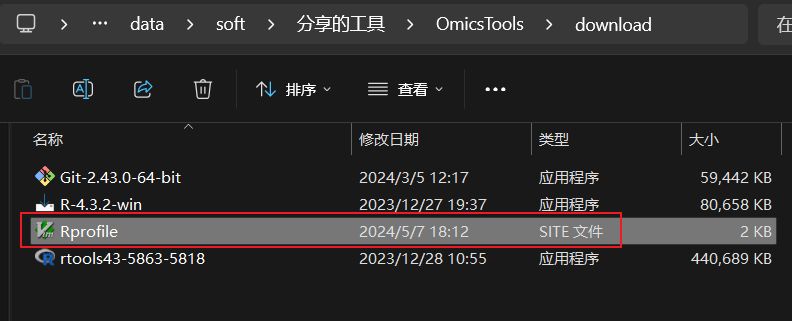
用我网盘的Rprofile.site替换掉原有的那个Rprofile.site
测试R语言分析环境的安装配置效果
打开cmd命令提示符

使用which命令在命令提示符终端中检索R和g++的启动路径
- 打开cmd命令提示符
- 打个which R,看看能否返回R的启动路径
- 打个which g++, 看看能否反馈g++的启动路径
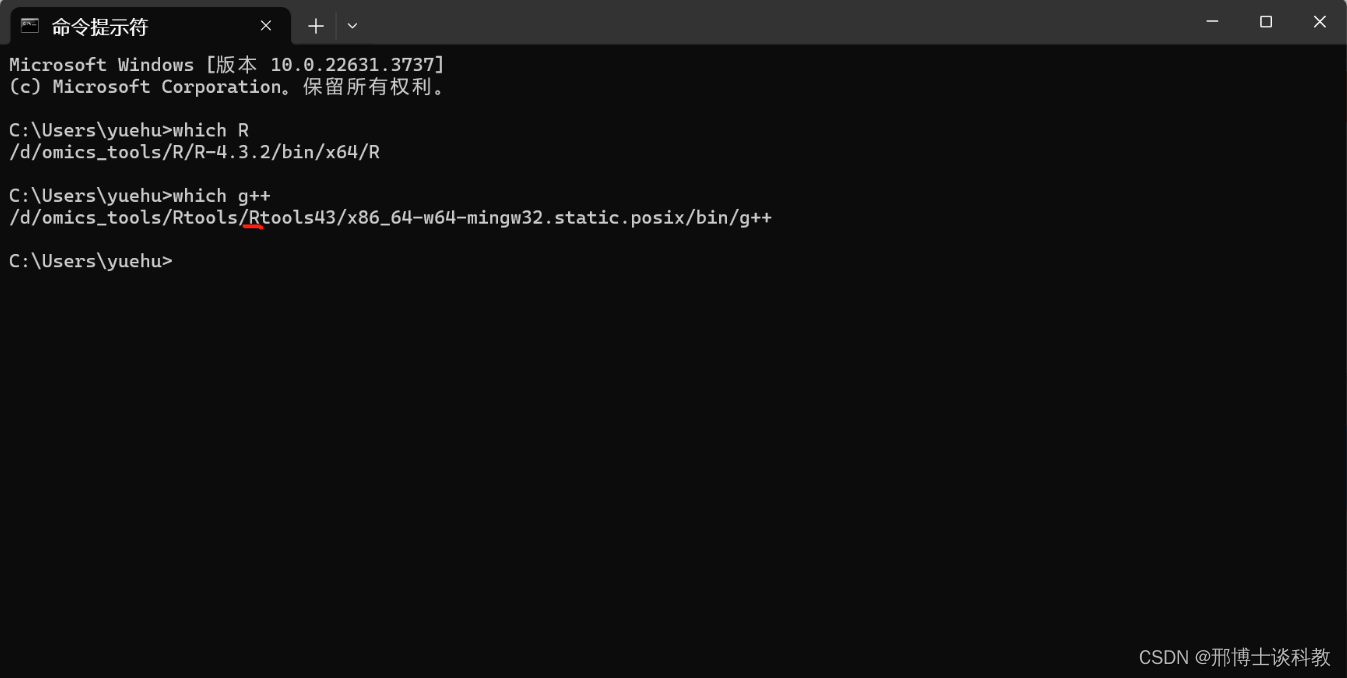
如果大家打开的cmd命令行没有返回路径,环境变量已经添加和保存的话,那可能是cmd终端打开的太早了,打开把原来的cmd终端关闭掉,再重新打开个新的cmd终端,来重新测试这些命令就行了。
如果能返回R的启动路径和g++的启动路径,就说明环境变量已经配置成功了。另外,如果返回的g++路径不是上图中的路径,很可能是大家看了生信自学网的课,下载了perl,并把perl的路径添加到系统变量里了,然后g++用的是perl里的g++, 大家也可以暂时不用管这个问题,因为g++在编译一些依赖c++的R包是很有用,但是所有的R包都已经被我编译适配好了,大家也就不用再管这个问题了。
或者大家也可以到系统环境变量里把perl的环境变量删除掉,我前面是让大家把R系列的环境变量添加再用户环境变量下了,perl的环境变量很可能是被大家添加到了系统环境变量下了,系统环境变量比用户环境变量全局性更强,同时环境变量是越靠前,越优先试用,要么大家也把Rtools系列的环境变量也添加到系统环境变量中,并移动到perl路径的上面,要么把系统环境变量中的perl路径给删掉。

测试R语言和R包能否正常工作
1. 打开cmd命令提示符,打个which R
2. 打个R, 启动R语言解释器
3. 打个library(Seurat), 测试一下R包的加载效果
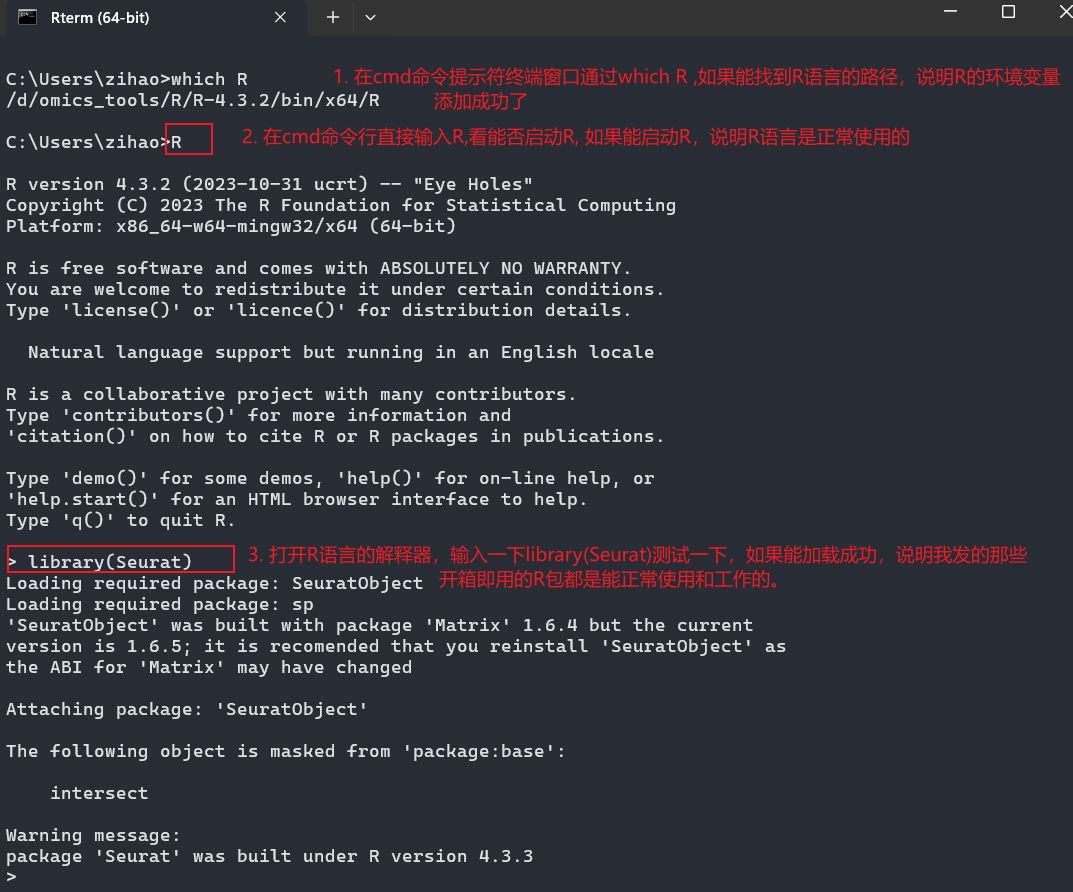
如果library(Seurat)能正常加载且没出现任何报错信息,就说明我给大家发的Seurat等大量开箱即用的R包都是能正常使用的。
至此,OmicsTools依赖的所有的R语言分析环境就完全配置完成了。
Seurat包加载报错的解决方法
解决方法是替换掉R语言原来的library下的所有R包
R语言安装目录下的library中也自带了一些R包,但是这些R包的版本有的比较旧了,尤其是Matrix包的版本比较旧,可能会导致单细胞分析的Seurat包和SeuratObject包加载失败报错的情况,在加载Seurat的时候会出现下面的这种报错:
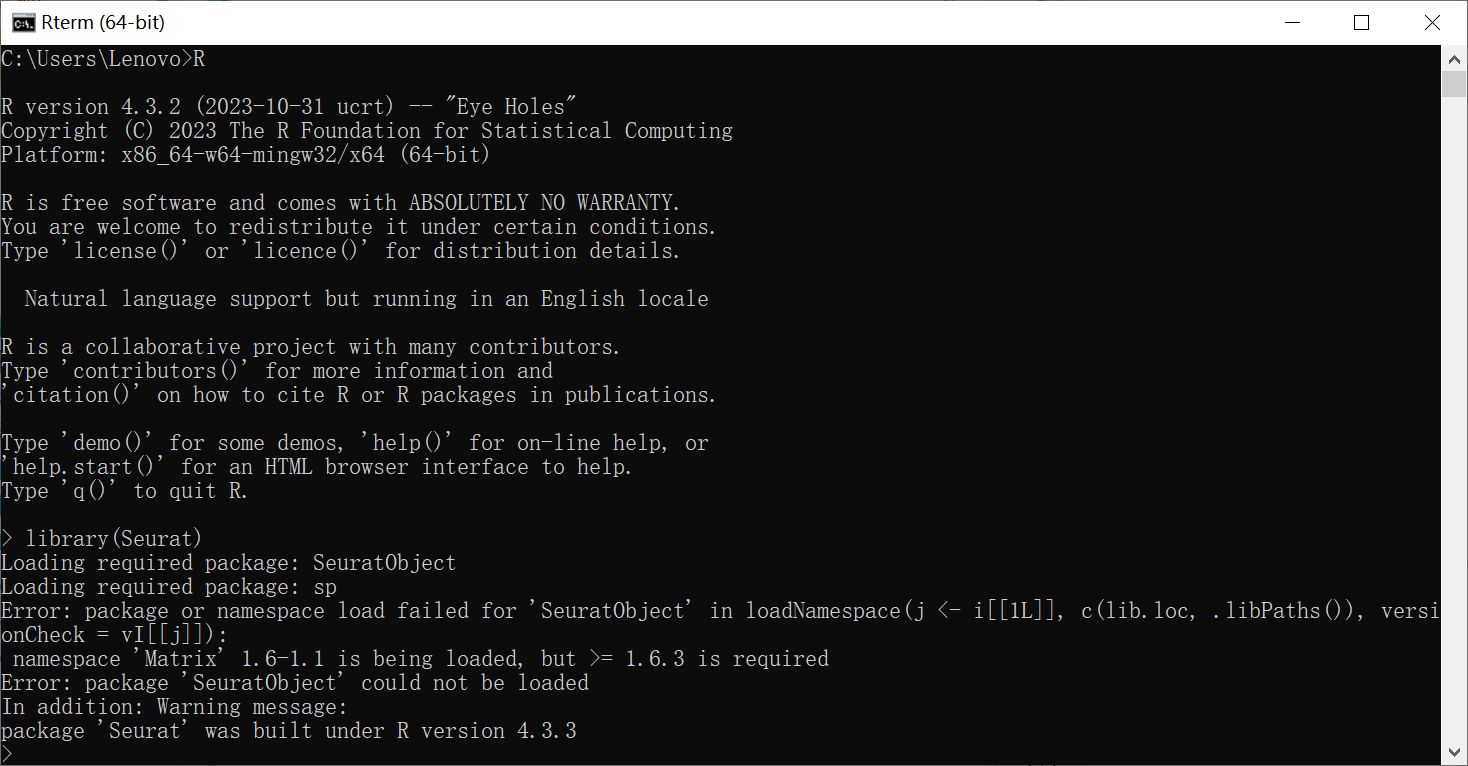
要替换的R包的网盘文件:

关闭cmd命令提示符,进入D:\omics_tools\R\R-4.3.2这个R语言的安装目录,删除library这个子目录
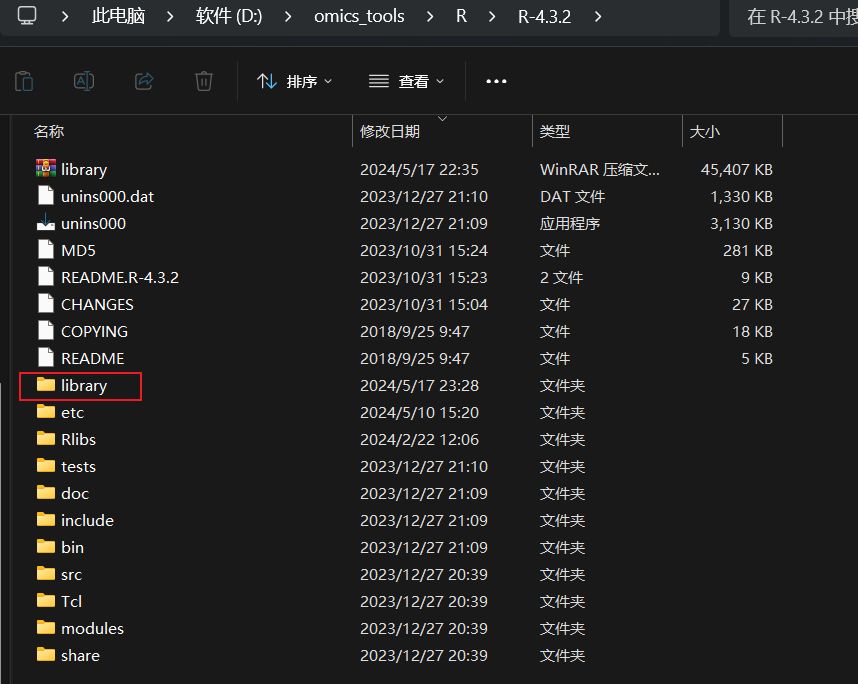
把我网盘里的library解压到D:\omics_tools\R\R-4.3.2下
解压好的library目录示意图如下:

替换工作完成后,再重新执行以下上一步的测试命令
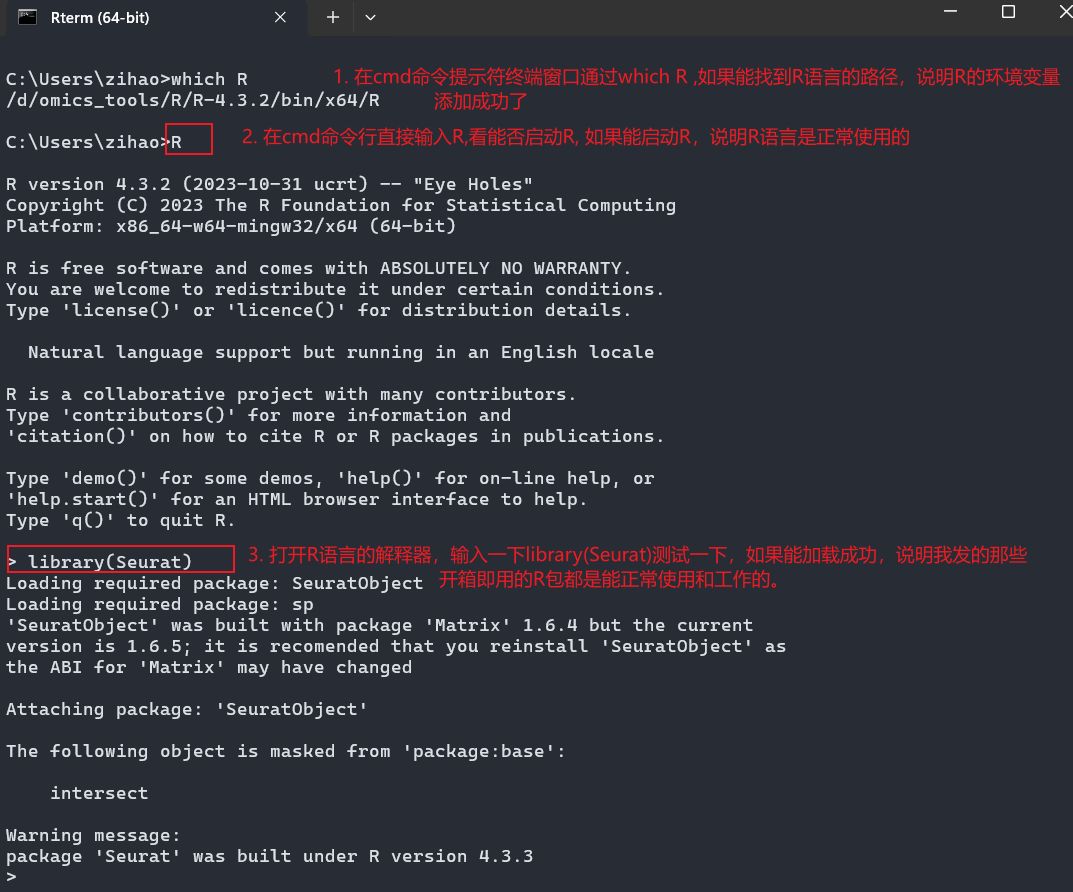
如果library(Seurat)能正常加载且没出现任何报错信息,就说明我给大家发的Seurat等大量开箱即用的R包都是能正常使用的。
至此,OmicsTools依赖的所有的R语言分析环境就完全配置完成了。
















 482
482











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











