







3D Infomax improves GNNs for Molecular Property Prediction
出处
- 作者:Hannes Stark等
- 机构:Massachusetts Institute of Technology等
- 期刊:Proceedings of the 39 th International Conference on Machine Learning,2022/06/04
- Code :github
摘要
- 使用现有的三维分子数据集来预先训练一个模型,以推理出仅有二维分子图的分子的几何形状。
- 模型的名称为 3D Infomax,最大化学习到的3D summary vector和GNN的表征之间的相互信息(mutual info)。
- 使用未知几何形状的分子进行微调,GNN仍然能够提供一些隐性的3D信息并用于下游任务。
- 在很多属性上有着较大的进步,比如在QM9量子力学特性上,MAE减少了22%
介绍
现有的分子特性预测方法和3D infomax的动机:
- 标准方法:利用GNN和2D的分子图,结果快但差;
- 显性的3D方法:使用经典的方法或者机器学习的方法计算3D坐标,然后作为输入进行预测。结果准确但是对于实际应用来说计算坐标太慢。
- 3D Infomax:① 预训练:用一个2D网络对有3D信息的分子进行训练,得到有着隐性3D信息的表征。② 将2D网络的参数微调。结果真是又快又好。
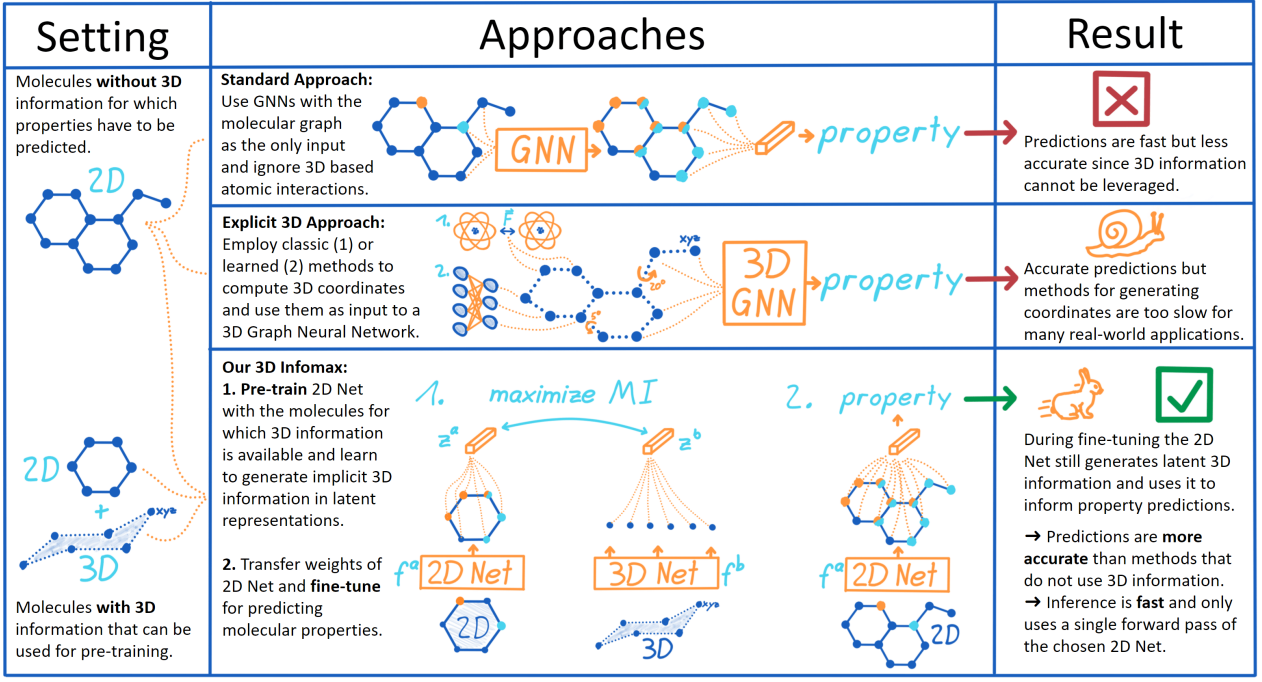
背景
- 2D分子图
- G = ( ν , ϵ ) ,其中 ν 是节点 − 原子, ϵ 是边 − 共价键 G=(\nu,\epsilon),其中\nu是节点-原子,\epsilon 是边-共价键 G=(ν,ϵ),其中ν是节点−原子,ϵ是边−共价键,边可以包含键类型信息,节点可以包含一些特征数据,比如原子编号,但至此都无3D坐标信息。
- 3D 分子构象
- 不同构象会带来不同化学性质,为保证抓住3D信息,所以需要考虑几乎所有的构象。
- 当考虑一个分子 c c c个已知的构象时,把他们表示成一组点云 { R } j 1 ⋯ c j \{R\}^j_{j1\cdots c} {R}j1⋯cj。每个点云 R = { r v } v ∈ ν R=\{r_v\}_{v \in \nu} R={rv}v∈ν表示分子中所有原子 ν \nu ν的坐标(即一组点云是一个构象的所有坐标点集合)。
- RD-Kit的ETKDG算法能快速计算构象但是不准确;最流行的是CREST,速度和准确率兼备,但仍然需要大约6小时(per cpu)完成一个药类分子的计算。
- 分子的对称性
- 当所有原子坐标 jointly translated或者围着一点旋转(SE(3)对称),那么分子的构象就不会改变。同时,分子的性质会被他们的手性决定。我们的方法也能在表征中体现对称性。
- 图神经网络
- 大部分GNN可以被一个MPNN框架描述,比如我们用的PNA模型。
- MPNN的目的是为了学习一个图的表征。他们通过不断迭代地去应用消息传递层,然后将所有点的表征结合。一个消息传递层通过使用置换不变性函数(mean,max,sum,不管数据如何置换位置,结果都不变)计算该点的邻点和其之间的边的值以用于更新该点信息。消息传递层之后,另一个置换不变性函数被用于提取点层的embedding到图层的embedding。
相关文献
- 分子属性预测
- MPNN框架问世之后,GNN就被广泛运用于量子化学、药物发现和分子性质的预测。利用3D信息的一个简单方法就是利用键长作为边信息(SchNet);DimeNet提取键角;SMP又包含另一个角度信息;GemNet也提取扭角,这样所有原子的相对位置就都被定义了;EGNN则是使用成对的原子距离。
- 自监督学习
- 对比学习是在计算机视觉中比较流行的自监督方法,通过对比相似的输入和不相似的输入的embedding来学习表征。
- 分子化学的数据集很小所以自监督学习很重要。不少研究者也利用对比学习来表征分子性质,但是很有限制也难以泛化。
- 过往大多是使用2D,我们和GraphMVP都是额外利用了3D结构来获取更多信息的表征。GraphMVP提出了一个生成式和一个对比式的3D预训练模型。生成模型可以纳入多个构象信息,然后利用对比式预训练提高下游任务表现。
- 我们与GraphMVP的区别:3D infomax不需要额外的生成预训练任务,直接在一个新的对比损失函数中包含了这些信息。此外,我们的评估包括量子力学任务,我们发现在这个领域可能的改进比非量子属性的改进要大得多。
方法(3D Infomax)
使用对比学习完成了输入为2D信息,但可以推断出3D几何信息的模型。预训练模型为PNA(将图多种方法聚合,sota和简单)
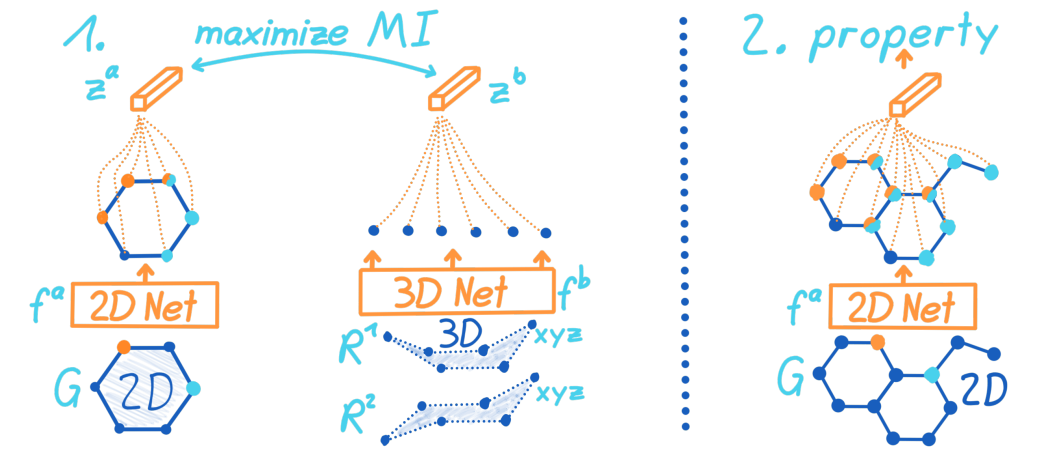
图1
- 最大化一个使用2D分子图的2D GNN和使用3D构象的3D GNN之间的互信息(在机器学习中,理想情况下,当互信息最大,可以认为从数据集中拟合出来的随机变量的概率分布与真实分布相同。)
- 与训练之后,我们将它迁移到属性预测任务,并微调。在微调过程中,GNN产出的3D信息会被用于提高预测。
- 在图1中,有两个模型。需要预训练的是2D网络
n
e
t
w
o
r
k
f
a
network f^a
networkfa,它可以产出一个表征
f
a
(
G
)
=
z
a
∈
R
d
z
f^a(G)=z^a \in \R^{d_z}
fa(G)=za∈Rdz;另一个将
R
=
{
r
v
}
v
∈
ν
R=\{r_v\}_{v \in \nu}
R={rv}v∈ν编码的3D网络
n
e
t
w
o
r
k
f
b
network f^b
networkfb给出一个表征
f
b
(
G
)
=
z
b
∈
R
d
z
f^b(G)=z^b \in \R^{d_z}
fb(G)=zb∈Rdz。可以当成是一个对比蒸馏,因为student 2D网络可以从teacher 3D网络那学会生产3D信息。
对比框架
为了教会2D n e t w o r k f a network f^a networkfa从2D图输入中学到3D信息,我们最大化了潜在2D表征 z a z^a za和3D表征 z b z^b zb的互信息。因为当两者来自同一个分子,那我们希望 z a z^a za和 z b z^b zb尽可能的一致,所以利用了图2的对比学习。
对于一组batch,中间包含N个分子图 { G i } i ∈ { 1 ⋯ N } \{G_i\}_{i \in \{1\cdots N\}} {Gi}i∈{1⋯N},点坐标 { R i } i ∈ { 1 ⋯ N } \{R_i\}_{i \in \{1\cdots N\}} {Ri}i∈{1⋯N},然后得到多个表征 z i a z_i^a zia和 z i b z_i^b zib。
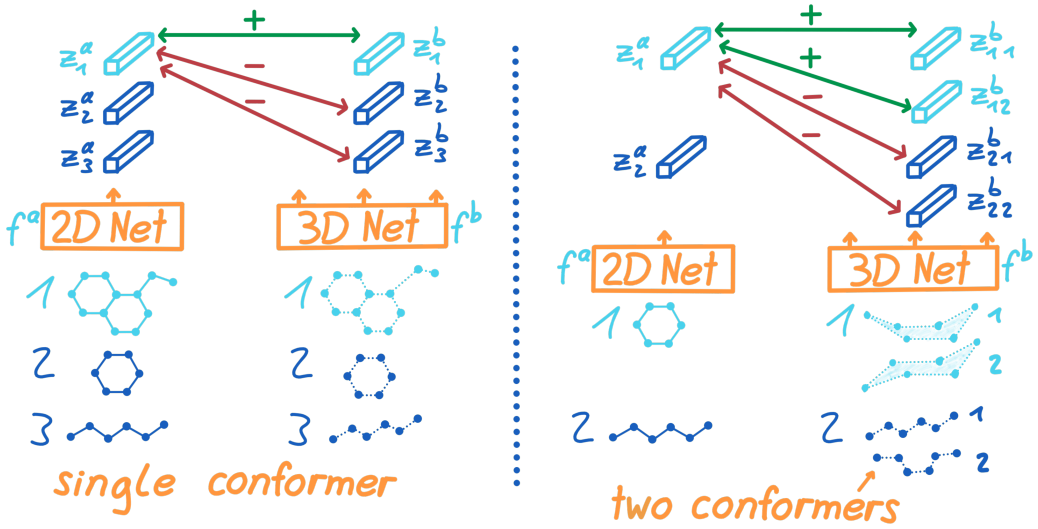 图2
图2
对比学习的第一个目的就是如果两者是正样本对,那么就要最大化表征的相似度,表示他们是来自同一个分子(同一个index i)。第二个目标就是强迫负样本对 z i a z_i^a zia和 z k b , i ≠ k z_k^b, i\ne k zkb,i=k不相似。
这两个目标都是通过修改NTXent loss实现(如何实现相似的越相似)的:
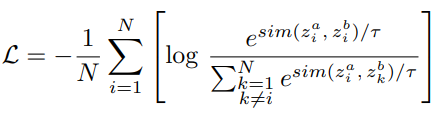
其中
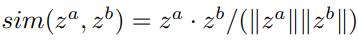
是余弦相似度, τ \tau τ是一个温度参数(超参),可以当做最相似的负样本对的权重(也就是当负样本对很相似时,调整 τ \tau τ)。不同的对比损失组合和自监督学习是有可能学会一个2D和3D表征之间的联合嵌入空间,上面的函数是表现最好的。
使用多构象
使用 c c c个最高概率的第 i 个构象 { R i j } j ∈ { 1 ⋯ c } \{R_i^j\}_{j \in \{1\cdots c\}} {Rij}j∈{1⋯c},如果不够c个就把能量最低的重复。图2右边,就是将分子的2D表征和每一个构象进行比较。
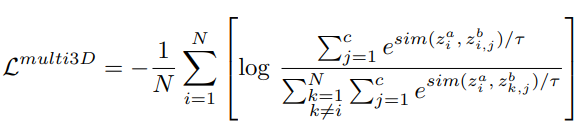
3D网络
3D网络输入是原子坐标作为点云,然后输出一个置换不变的表征
z
b
z^b
zb,尽可能多的把3D结构信息编码,但是不能够接触2D信息,不然的话互信息可能会因为两个模型的交互变得更大。
我们的模型将每一对atom的欧几里得距离进行编码,这样表征可以定义所有原子的相对位置并且保证旋转平移不变性,并且也是反射不变的,但是对手性分子没办法区别。
d
u
v
d_{uv}
duv表示u原子和v原子之间的距离,会先使用高频的sine和cosine去投影到一个高维空间(因为键长之间的区别比较小)。然后以F=4的频率map(有点类似position encoding),更详细的操作可见MPNN框架。
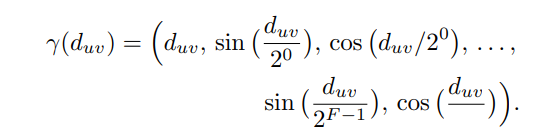
数据
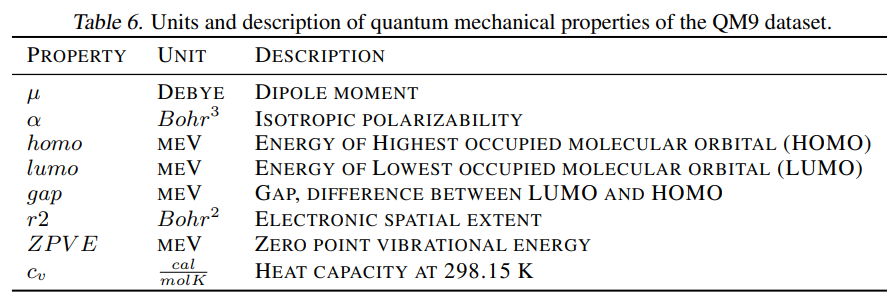
- 3D数据集是来自QM9(134k个平均18个原子的只有一个构象的小分子,kaggle下载)、GEOM-Drugs(304k)和QMugs(665k)。后两者有较大的且是多构象的药类分子(44.4和30.6平均原子个数)
- 微调:预测十个来自QM9和GEOM-Drug的量子特性,这些数据不与预训练的数据相交。
- 预训练用了50k单构象来自QM9,140k5构象来自GEON-Drugs,620k3构象来自QMugs
对比
Baseline
- 距离预测:使用已有的最低能量构象去预训练一个GNN,以直接预测所有原子之间的距离。然后将任意两个u,v原子的表征简单地拼接在一起(uv,vu),随后放入mlp(U,直接降到1维),||表示拼接
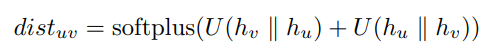
s o f t p l u s ( x ) = l o g ( 1 + e x ) softplus(x) = log(1 + e^x ) softplus(x)=log(1+ex),loss function是MSE。 - 构象生成:GeoMol(sota生成分子构象的模型),一个生成式模型,产生一个分子的可能的3D结构的分布,从而获取到多构象信息。利用他们模型做预训练任务然后提取网络用于不同下游任务
- GraphCL:一个卷积增强预训练模型with JOAO配置,模型通过学习产出一个对增强不变的表征来完成自监督目标。
结果
数值为MAE,RAND INIT模型随机初始参数,PROPRED指用GEOM-Drugs的Gibbs自由能来做的预训练,DISPRED指用有最高概率的构象去预测所有原子的距离,CONFGEN指与训练的时候预测10个构象,3D Infomax分别使用三个数据集做预训练,RDKIT SMP使用RDKit生成的3D坐标输入SMP(一个GNN)做训练,True 3D SMP最后一列是用真实的3d坐标使用SMP预测的,蓝色表示improvement,橙色表示worse。
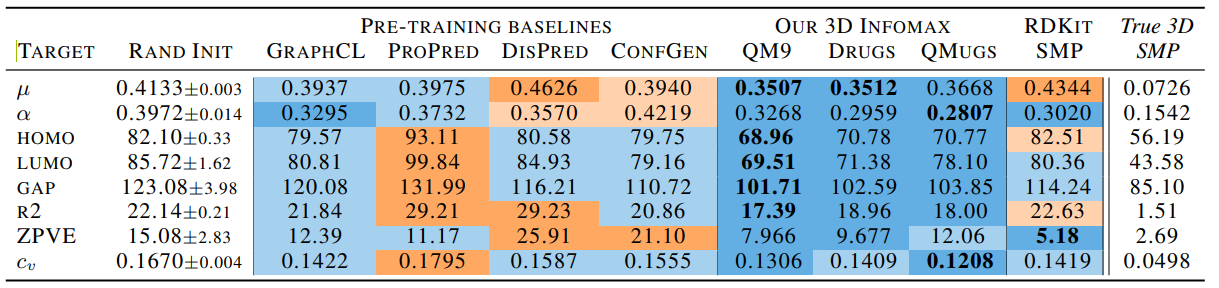
对QM9数据集中的8中特性做预测
结论
相当于一个2D分子图的预训练模型,能得到隐含3D信息的表征,并且具有一定泛化能力(不会有负迁移),可以借助同一个分子的多构象信息来帮助下游属性预测任务。

















 2484
2484

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









