






 该论文提出了一种名为GeoGNN的新型图神经网络,结合了分子的拓扑和几何信息,以改进分子表征学习。通过自监督学习策略,包括局部和全局空间结构信息的预测,GeoGNN在多个分子属性预测数据集上表现出优越性能。这种方法强调了分子的3D几何结构在化学特性预测中的重要性。
该论文提出了一种名为GeoGNN的新型图神经网络,结合了分子的拓扑和几何信息,以改进分子表征学习。通过自监督学习策略,包括局部和全局空间结构信息的预测,GeoGNN在多个分子属性预测数据集上表现出优越性能。这种方法强调了分子的3D几何结构在化学特性预测中的重要性。
基本信息
这是一篇由百度公司的研究人员于2021年发表在计算机领域顶刊nature子刊nature machine intelligence(Nat Mach Intell) 上的一篇论文,关键贡献在于将三维空间几何信息考虑到了分子表示学习当中。
博客创建者
武松
作者
Fang, Xiaomin and Liu, Lihang and Lei, Jieqiong and He, Donglong and Zhang, Shanzhuo and Zhou, Jingbo and Wang, Fan and Wu, Hua and Wang, Haifeng
标签
深度学习,图神经网络,自监督学习,分子表征学习,分子属性预测
摘要
有效的分子表征学习对于促进分子性质预测至关重要,这是药物和材料行业的一项基本任务。图神经网络(GNNs)的最新进展在将GNNs应用于分子表征学习方面显示出巨大的前景。此外,最近的一些研究也证明了自监督学习方法在预训练GNN以克服标记分子不足的问题方面的成功应用。然而,现有的GNN和预训练策略通常将分子视为拓扑图数据,而没有充分利用分子几何信息。然而,分子的三维(3D)空间结构,即:分子几何结构,是决定分子物理、化学和生物特性的最关键因素之一。为此,作者提出了一种新的用于化学表征学习(ChemRL)的几何增强分子表征学习方法(GEM)。该方法主要包含以下两个贡献:
- 提出了一种基于几何信息的图神经网络GeoGNN,来编码分子的拓扑和几何信息;
- 引入了多个几何级的自监督学习任务来学习分子的三维空间知识;
作者在各种分子性质预测数据集上对ChemRL-GEM进行了彻底的评估。实验结果表明,ChemRL-GEM在多个基准上显著优于基准方法。
问题定义
- 基于图的分子表示
基于图的分子表示即将分子看作一个图,使用一系列方法将分子的信息表示成可计算处理的形式,如矩阵、向量。近年来的方法主要包括三类:基于分子指纹,基于线性序列,基于图神经网络。基于分子指纹的如ECFP、MACCS,基于线性序列的如SMILES串,基于图神经网络的方法例如GCN、GIN、GAT等,通过聚合更新节点和邻居信息来获得分子表示。
- GNNs的预训练方法
在分子表征学习领域,最近的一些研究探索了自监督学习对提高GNN模型在下游任务上的泛化性能的能力。这些工作主要关注两种自监督学习任务:节点级(边级)任务和图级任务。设计了节点级自监督学习任务来获取局部域知识。例如,一些研究随机掩码节点或子图的一部分,然后通过节点/边表示来预测它们的属性。图级的自监督学习任务用于捕获全局信息,比如通过图表示来预测图的属性。通常,图的性质是特定领域的知识,如从生化分析的实验结果或分子官能团的存在。
方法
方法架构图
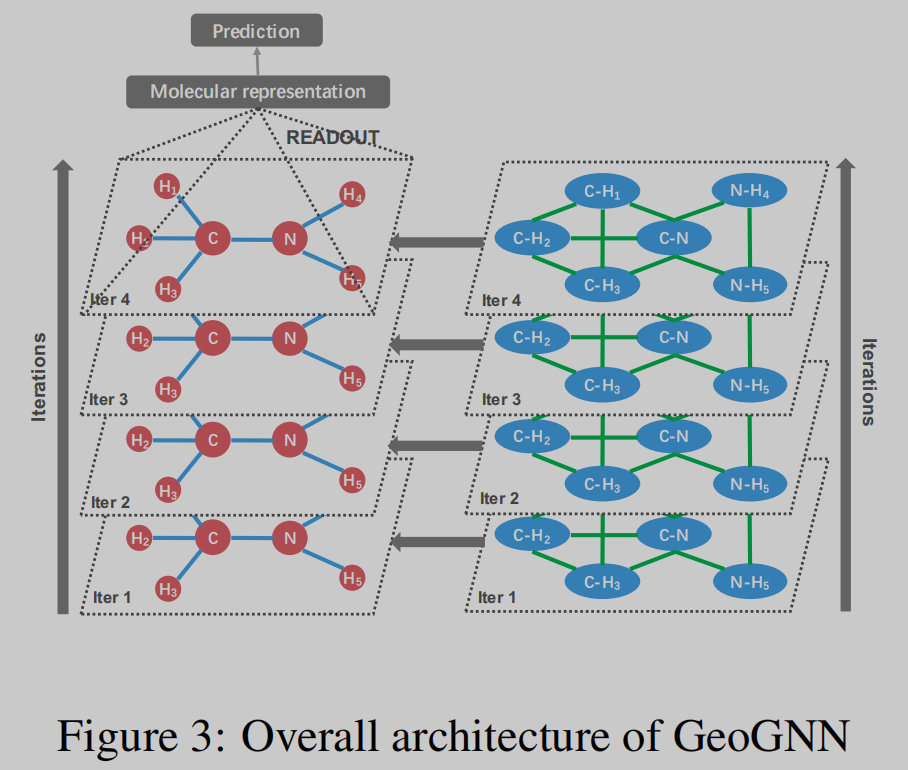
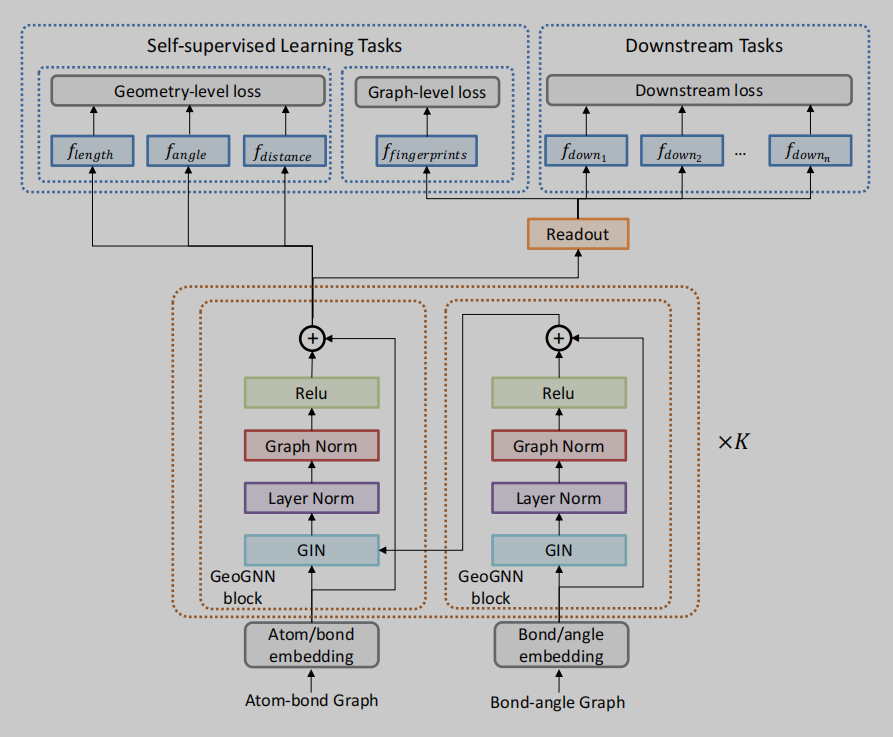
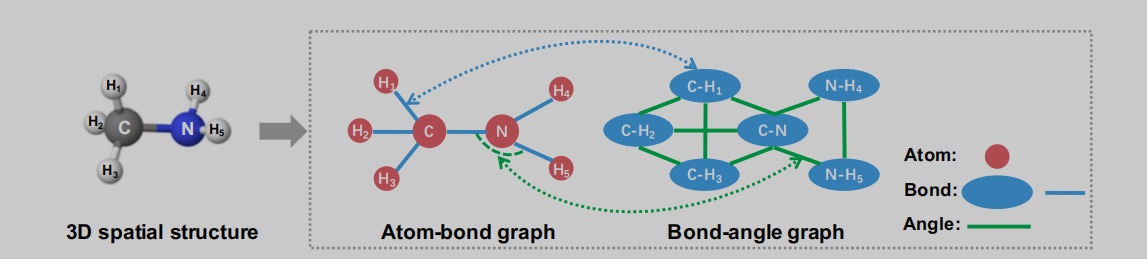
方法描述
GeoGNN:基于几何信息的GNN
GeoGNN将几何信息融合考虑,主要体现在对原子信息、键信息和键角信息的融合考虑。如图2所示,作者将网络分成两个部分,即:atom-bond和bond-angle。如图1所示,左半部分即atom-bond网络,用于提取原子和键的信息,右半部分即bond-angle,用于得到键角的信息。网络的backbone使用的是GIN,添加了常见的layer norm等技巧堆叠而成GeoGNN block。由两部分图网络分别进行特征提取。不同于一般的GNN,GeoGNN的消息传递过程有所不同,聚合和更新操作分为三部分进行。如公式1所示,考虑中心原子相关的某一条键,先由键的特征和与这条键相关的键角特征进行聚合,其中 x w u v x_{wuv} xwuv表示由键wu和键uv组成的键角特征。
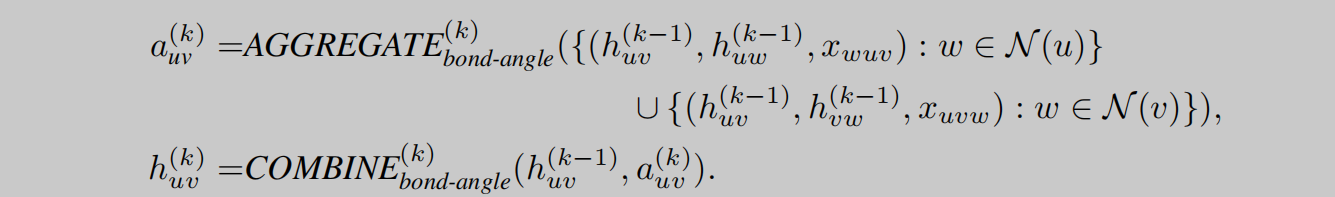
如公式2所示,在得到中心原子键的特征后,再对每个相邻的键进行聚合,得到聚合特征 a u ( k ) a^{(k)}_u au(k),此时的 a u ( k ) a^{(k)}_u au(k)已经包含了键角信息了,接下来只需要再将分子图中的各个原子更新后的特征进行聚合即可获得分子表示,即readout操作,如公式3所示。
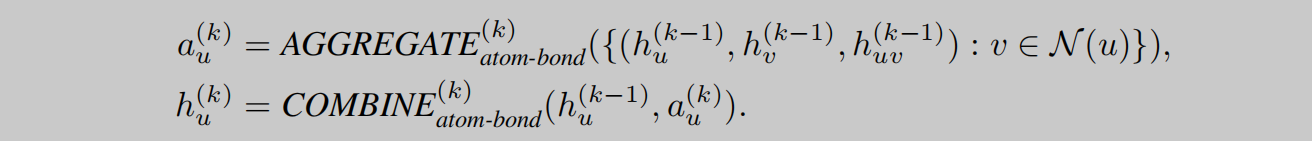
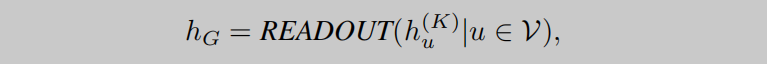
考虑几何信息的自监督训练策略
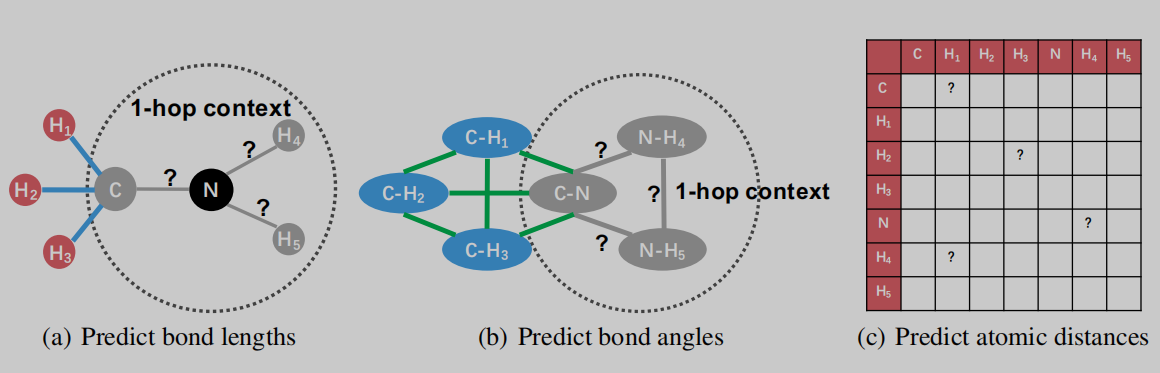
本文的预训练分为两个部分,一个是几何级别的训练和图级别的训练。前者即下面要介绍的局部空间和全局空间结构信息的训练,是将GeoGNN Block得到的节点表征直接用来进行预测的;后者是用得到的节点表示readout后得到的图级表示来预测分子指纹MACCS和ECFP。
- 局部空间结构信息
局部空间结构信息的训练通过如图4(a)、(b)所示来进行,遮盖1-hop范围内的(mask)原子信息、键信息、键角信息,建立预测值与遮盖值之间的Loss函数,通过自监督训练预测遮盖部分,完成局部空间信息的训练。图4(a)表示进行键长的预测,(b)表示进行键角的预测。在公式4中, f f f表示模型预测值, l u v l_{uv} luv和 ϕ u v w \phi_{uvw} ϕuvw表示ground truth。
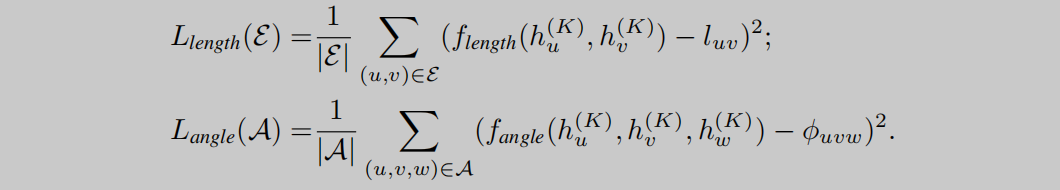
- 全局空间结构信息
全局空间信息的学习需要模型预测出分子内部每两个原子之间的距离,这个距离以及前面的键长都是通过RDKit提供的函数得到的模拟3D坐标来计算的。如公式5所示,作者使用 b i n bin bin函数来将标准值 d u v d_{uv} duv转换成one-hot向量,再与预测值 f d i s t a n c e f_{distance} fdistance相乘。
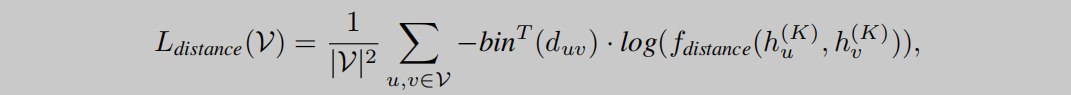
最终以局部和全局的3个损失函数相加作为整个与训练过程的损失函数。
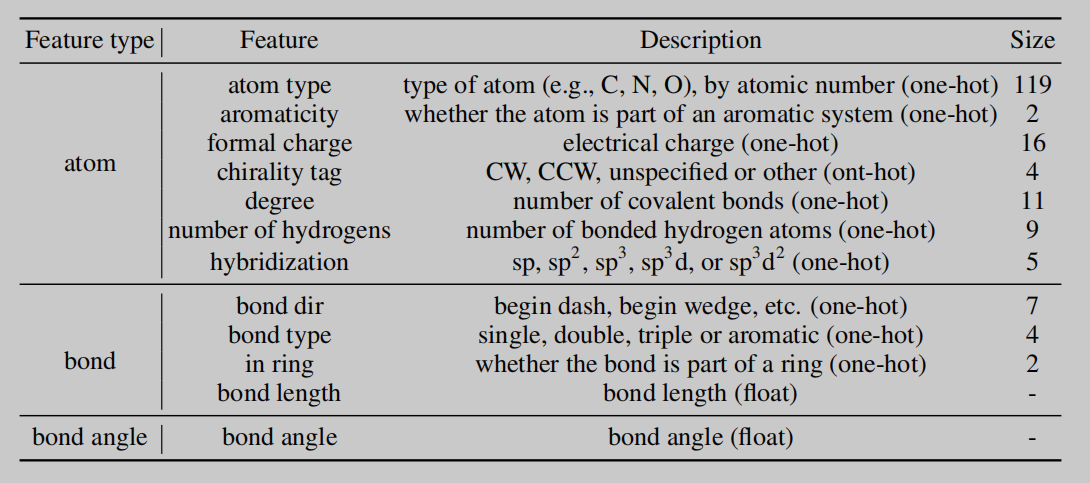
输入特征
输入特征的详细参数也是由RDkit的函数直接或者间接得到的。
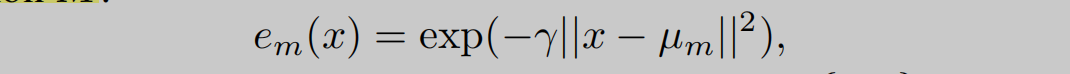
其中,
μ
m
\mu_m
μm即一个步长为0.1的数组,与x分别作差平方后,得到一个新的数组,然后再乘上参数
γ
\gamma
γ从而得到一个关于原来标量值的一个向量表示,具体思想参考径向基函数
(Radial Basis Functions)。
实验
实验设置
数据集
- 训练数据集
ZINC15:最大的有机小分子化合物数据库之一 - 分子属性预测数据集
MoleculeNet:专门设计用于测试分子特性的机器学习方法的基准数据集,内含有多个分子数据集
参数设置
这篇文章实验对所有模型使用Adam优化器,学习率为0.001。对于每个数据集,训练了批处理大小为32的模型。根据moleculeNet的建议,使用平均ROC-AUC作为6个二值分类数据集的评价度量。对于回归数据集,对于FreeSolv、ESOL 和Lipo,使用均方根误差(RMSE),而对于QM7、QM8和QM9 ,使用平均平均误差(MAE)。对每种方法执行4次独立的运行,并报告这些指标的平均值和标准偏差
基准模型
- D-MPNN
- AttentiveFP
- N-Gram
- PretrainGNN
- GROVER
实验结果及分析
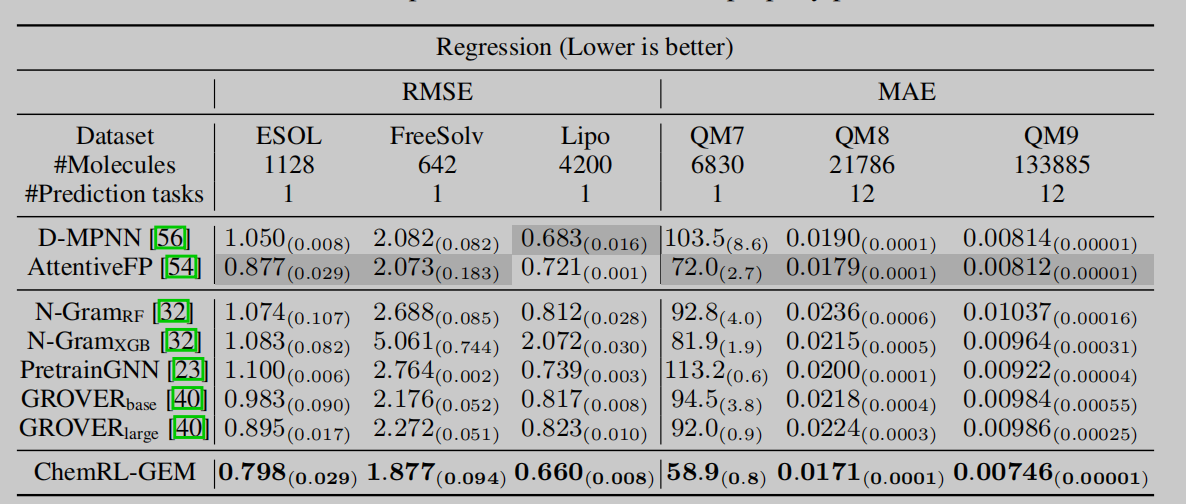
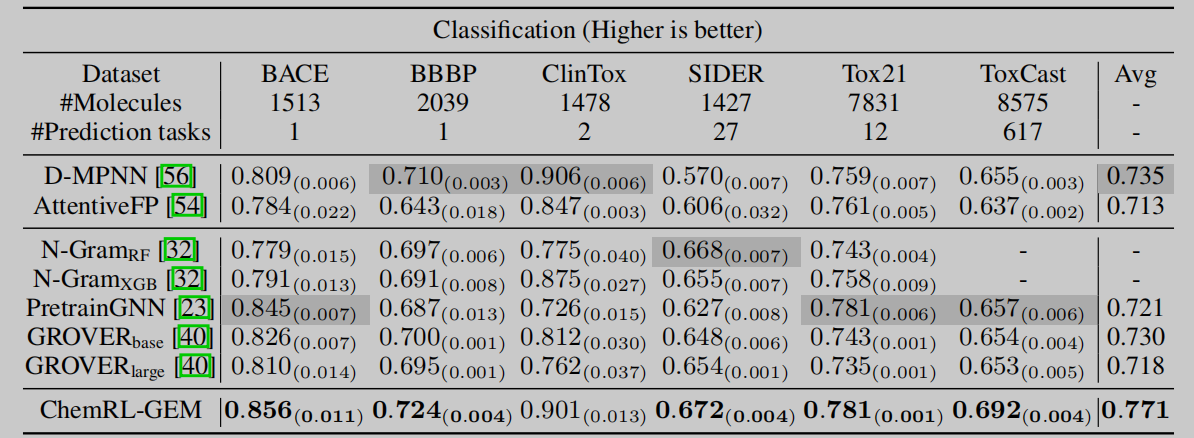
作者分别在分子属性预测相关的一共12个分类和回归数据集上进行实验,数据集来自MolecularNet,并进行了不同的分子属性预测任务。在两类数据集上,作者所提出的模型都取得了最好的效果。
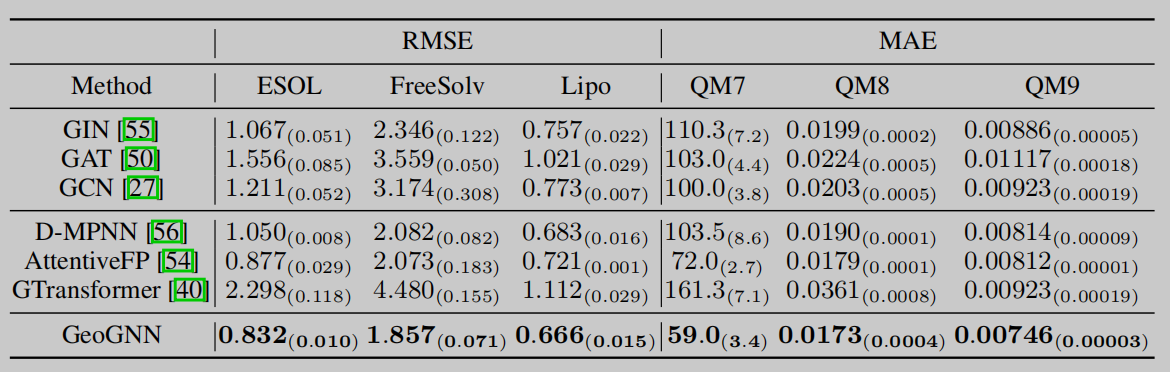
消融实验
作者设计了消融实验来验证模型两个部分的有效性,分别是验证GeoGNN的作用和验证设计的基于几何信息的预训练策略的有效性。这里有疑惑的一点就在于这里GeoGNN验证部分,论文中看起来仅仅是进行了与其他基准模型的对比,然而本应该是取消GeoGNN的对几何信息的考虑,来看几何信息对性能的贡献才对。
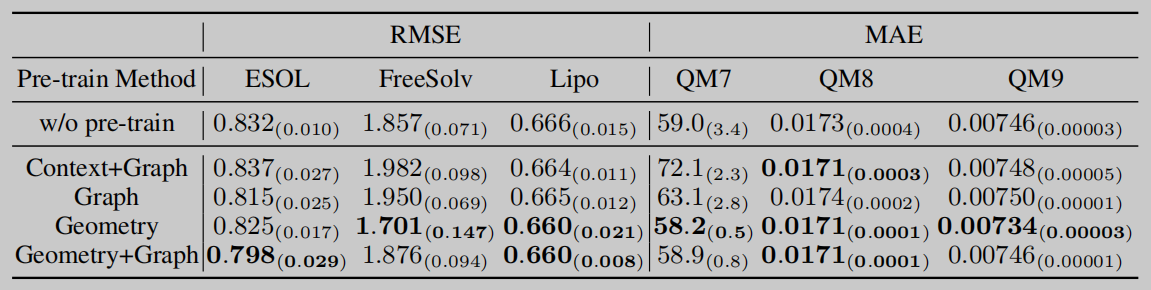
相关知识链接
- Yang K, Swanson K, Jin W, et al. Analyzing learned molecular representations for property prediction[J]. Journal of chemical information and modeling, 2019, 59(8): 3370-3388.
- Xiong Z, Wang D, Liu X, et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism[J]. Journal of medicinal chemistry, 2019, 63(16): 8749-8760.
- Liu S, Demirel M F, Liang Y. N-gram graph: Simple unsupervised representation for graphs, with applications to molecules[J]. Advances in neural information processing systems, 2019, 32.
- Hu W, Liu B, Gomes J, et al. Strategies for pre-training graph neural networks[J]. arXiv preprint arXiv:1905.12265, 2019.
- Rong Y, Bian Y, Xu T, et al. Self-supervised graph transformer on large-scale molecular data[J]. Advances in Neural Information Processing Systems, 2020, 33: 12559-12571.
下载
基准实验涉及的论文
亮点
- 创造性地将几何信息考虑到分子表示学习之中
[不足]
- 消融实验中验证考虑几何信息的GNN的有效性实验部分,设计的不够合理
[启发]
- 对几何信息的考虑,可以应用到其他的下游任务中直接做端到端训练,比如DDI任务中,是否可以将几何信息直接构建到相互作用模块中
BibTex
@article{fang2021chemrl,
title={Chemrl-gem: Geometry enhanced molecular representation learning for property prediction},
author={Fang, Xiaomin and Liu, Lihang and Lei, Jieqiong and He, Donglong and Zhang, Shanzhuo and Zhou, Jingbo and Wang, Fan and Wu, Hua and Wang, Haifeng},
journal={arXiv preprint arXiv:2106.06130},
year={2021}
}

















 530
530

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









